www.earticle.net
DNA Degradation이 Genotyping-by-Sequencing(GBS) 자료 기반 한우 유전적 표지 발굴에 미치는 영향
유동안1 · 김권도1,2 · 김덕경2 · 조서애2*
서울대학교 생물정보학 협동과정1, (주)조앤김지노믹스2
Effects of DNA Degradation on the Genotyping-by-Sequencing (GBS) Data for Genetic Marker Discovery of Hanwoo Population
Dong Ahn Yoo1, Kwondo Kim1,2, Duk Kyung Kim2 and Seoae Cho2*
1Interdisciplinary Program in Bioinformatics, Seoul National University, Seoul 08826, Korea
2C&K Genomics Inc., C-1008, H Businesspark, 26, Seoul 05836, Korea
ABSTRACT1)
Genotyping-by-sequencing (GBS) is a cost-efficient method which can be useful for SNP marker discovery in a population of interest. GBS is genome reduction sequencing method using restriction enzyme. The quality of DNA is a key factor which could have an influence in downstream analysis. However, there have not been many studies which investigated the impact of DNA degradation and the quality of the data on marker discovery. In this study, GBS data of 6 Hanwoo samples (H1~6) showing differing level of DNA degradation were compared. Re-sequencing pipeline was followed to investigate the impact of DNA degradation on marker discovery. As a result, we found that the quantity and quality of SNPs were not affected in the sample H5 and H6 with moderately degraded DNA. On the other hand, marker discovery was greatly affected in samples with severe DNA degradation (H3 and H4). The findings in this study support that GBS is a robust genotyping method towards moderate DNA degradation.
(Key words: Hanwoo, GBS, DNA degradation, DNA marker, Genotyping)
Ⅰ. 서론
GBS(genotype-by-sequencing) 기술은 유전체에서 특정 제한 효소의 인식 부위(recognition site) 주변을 sequencing 하는 기술이다. GBS 기술은 먼저 유전체를 대상으로 제한 효소를 처리하여 유전체를 잘라내고, DNA단편을 이용, sequencing library를 제작하여 제한 효소의 인식부위 주변 부분의 염기서열을 결정한다. 일반적으로 제한 효소의 인 식부위는 종 내의 각 개체 유전체 상에서 보존되므로
sequencing 된 인식부위 주변 염기서열은 유전적 표지 (genetic marker) 로서 유용하게 사용될 수 있다. GBS기술 은 전장 유전체 sequencing(whole genome sequencing)에 비해 유전체의 일부분만을 대상으로 하기 때문에 소요되는 비용이 훨씬 적다(De Donato et al., 2013). 따라서 GBS기 술을 이용하면 적은 비용으로 전장 유전체 sequencing 에 비해 많은 수의 샘플을 포함한 집단의 연구가 가능하다 (Elshire et al., 2011, He et al., 2014). GBS와 같은 sequencing기반 연구에 있어서 DNA degradation 정도나
* Corresponding author: Seoae Cho, C&K Genomics Inc., Seoul 05836, Korea, Tel: +82-2-876-8820, E-mail: [email protected] This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc-nd/3.0/deed.ko), which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited. The moral rights of the named author(s) have been asserted.
[Provider:earticle] Download by IP 118.70.52.165 at Monday, December 20, 2021 7:51 PM
www.earticle.net
DNA의 품질은 매우 중요한 요소이다. 예를 들어, DNA 품 질 문제 중의 하나인 cytosine의 deamination은 sequencing 에서 실제와 다른 염기서열을 생산할 수 있다(Brotherton et al., 2007). 또한 DNA 변성은 DNA의 손상에 의해 염기 를 잘못 읽는 DDML(damage-derived miscoding lesion) 현상을 일으킬 수 있으며, 염기의 변형은 DNA의 증폭 과 정을 중단 시킬 수도 있다(Friedberg et al., 2005). 특히 집 단 유전체의 유사한 부분을 sequencing 해야 하는 GBS기 술에서 제한효소 인식부위 및 그 주변 염기서열이 절단 또 는 변성에 의해 영향을 받은 경우, 이러한 유사한 부분들 을 발견하고, 유전자형을 추정하는 과정이 어려워 질 수 있다. 또한 DNA degradation에 의한 염기서열의 절단은 GBS에 의해 sequencing 되는 DNA 단편서열을 조각화 시 켜 정보의 손실을 일으킬 수 있으며(Graham et al., 2015), 전장 유전체의 일부분만을 이용하는 GBS 기술의 특성상 이러한 정보의 손실은 후속 분석결과에 큰 왜곡을 일으킬 지도 모른다.
DNA degradation은 여러 가지 원인에 의해 일어날 수 있다. 예를 들어 DNA를 다루는 과정에 생기는 물리적인 힘에 의해 fragmentation, nicks가 생길 수 있고, DNA가 오랜 기간 습한 환경에 저장된 경우나 열, 자외선, pH의 변화, 염분 등에 노출된 경우에는 분해될 수도 있다 (Lindahl et al., 1972, Lindahl 1993, Bruskov et al., 2002, Cadet et al., 2005). 죽은 세포나 다양한 환경에서 채취한 DNA에서도 이러한 여러 가지 원인에 의해서 cytosine deamination을 포함한 여러 종류의 변성 및 절단이 자주 일어난다(Hofreiter et al., 2001). 이렇게 다양한 원인으로 인해 발생하는 DNA degradation 은 GBS 를 통해 생산한 자료의 품질에 결정적인 영향을 미칠 수 있다. 그럼에도 불구하고 아직까지 DNA degradation이 GBS 과정 및 생 산된 자료에 미치는 영향에 대한 연구는 거의 이루어지지 않았다(Graham et al., 2015).
본 연구는 GBS 기술을 이용하여 생산한 한우 유전체 염 기서열 자료 중 품질 차이를 보이는 한우 6두를 선택하여, 생산된 자료의 양적 및 질적 비교를 통해 DNA degradation 이 GBS 자료 생산에 미치는 영향을 분석하였다. 또한, 이 를 통해 GBS 기술 기반 유전적 표지 발굴에 사용하기 적 합한 DNA의 품질을 밝히고자 하였다.
Ⅱ. 재료 및 방법
1. Genotype-by-sequencing 을 통한 단편서열 생산
GBS 분석에 앞서 연구에 사용할 한우 시료를 선별하기 위하여 한우 시료들을 대상으로 agarose gel 전기영동을 수행하였다. 전기영동을 통하여 DNA 품질을 평가하였으 며 이 정보를 바탕으로 한우 시료 H1~6를 선발하였다. 이 때 H1, H2는 거의 온전한 DNA를 가진 시료였으며, H3, H4 는 가장 높은 수준으로 DNA degradation 이 진행된 시료, H5, H6는 앞선 두 그룹의 중간 정도의 품질을 가진 시료였다.
GBS를 위한 DNA library는 앞선 연구(Elshire et al., 2011, De Donato et al., 2013)와 비슷한 과정을 거쳐 준비 하였다. 먼저 DNA를 Pst I 제한 효소를 이용하여 잘라내 었으며 adapter 서열과 ligation 시켰다. Ligation은 T4 DNA ligase(New England Biolabs, USA)를 이용하여 22℃
에 2시간동안 이루어졌으며 그 이후 ligase의 불활성화를 위하여 65℃ 에 20분의 배양과정을 수행하였다. 이때 adapter 서열은 index 서열을 포함하여 시료 별 구분을 할 수 있도록 하였다.
Adapter 와 ligation 된 총 6 샘플의 한우 DNA를 혼합 하고, 이를 quick PCR purification kit(Qiagen, Germany) 을 이용하여 정제하였다. 정제된 DNA의 0.1μg 을 대상으 로 50μl의 AccuPower Pfu PCR premix(Bioneer, Daejeon, South Korea), 25pmol 의 forward, reverse primer 를 이 용하여 PCR을 수행하였다. Primer의 서열은 다음과 같다:
5’-AATGATACGGCGACCACCGAGATCTACACTCTTTC CCTACACGACGCTCTTCCGATCT -3’, 5’- CAAGCAGA AGACGGCATACGAGATCGGTCTCGGCATTCCTGCTG AACCGCTCTTCCGATCT -3’.
앞서 준비한 혼합물을 98℃에 5분동안 둔 후, 18 cycle의 PCR을 수행하였다. 이 때 한 PCR cycle은 98℃ 에서 10초, 65℃ - 5초, 72℃ - 5초로 이루어진다. PCR의 결과물은 QIAquick PCR purification kit(Qiagen)을 이용하여 정제 하였고, 증폭된 DNA 조각의 크기 분포는 BioAnalyzer 2100(Agilent Technologies, Santa Clara, CA, USA)를 이용 하여 시각화 하였다. GBS library는 Illumina NextSeq500(Illumina, San Diego, CA, USA)를 이용하여 염기서열 분석을 진행하였다.
2. 단편염기서열 자료 정렬을 통한 SNP발굴
하나의 자료로 생산된 염기서열을 각 시료 별로 구분하 기 위해, index 서열과 GBSX(Herten et al., 2015) 프로그램
[Provider:earticle] Download by IP 118.70.52.165 at Monday, December 20, 2021 7:51 PM
www.earticle.net
을 이용, de-multiplexing을 수행하였다. 각 시료 별로 나 누어진 염기서열 자료를 대상으로 Trimmomatic 프로그램 (Bolger et al., 2014)을 이용하여 adapter 서열 및 품질점수 (quality score)가 낮은 부분(quality score<30), 전체 길이 가 짧은 서열(base pair<75)을 제거하였다. 이를 참조 유전 체 서열(UMD3.1) 에 Bowtie2 프로그램을 이용하여 정렬 시킨 후(Langmead et al., 2012), Picard프로그램을 이용하 여 한번의 자료 생산에서 생성된 서열을 모아주는 read group 추가 과정을(http://picard.sourceforge.net) 거쳤다.
추가적으로 duplicate 제거, local realignment, base quality recalibration 과정을 수행하여 re-sequencing 과정 에서 생길 수 있는 오류를 최소화 하였다. GATK(Genome Analysis Toolkit) 프로그램(McKenna et al., 2010)을 이용 하여 정렬이 완료된 시료들의 변이 정보를 추출하였으며, 최종적으로 다음과 같은 조건에 맞는 변이를 추출하였다: 1) MQ0(mapping quality zero)>=4, 2) QUAL(quality score)<30, 3) QD(quality by depth)<5.0, 4) FS(Fisher strand)>200.
Ⅲ. 결과 및 고찰
1. Agarose gel 전기영동을 통한 DNA 품질 비교
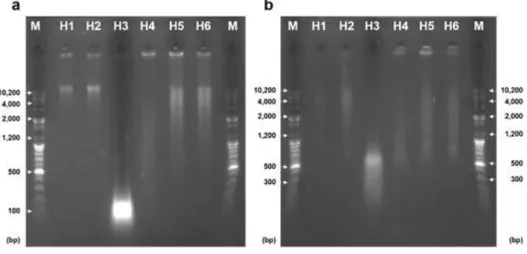
DNA 품질 비교에 대한 결과는 Fig. 1a와 같다. DNA 품 질을 수치화 하기 위하여 ~10,000bp 이하의 길이를 가진 DNA를 대상으로 DNA 양을 정량화하였다(Fig. 2). 그 결
과 DNA의 품질이 높고 degradation이 거의 진행되지 않 은 H1, H2시료는 대부분의 DNA가 10,000bp 주변에 분포 하였고, Fig. 1a 에서 보듯이 대부분의 DNA가 agarose gel 을 얼마 이동하지 못한 것을 관찰할 수 있었다. 그에 비해 H3 과 H4시료는 DNA degradation의 정도가 심하여 DNA의 가장 높은 peak가 35bp 주변에 분포하거나 35~10,000bp에 전체적으로 분포하였으며, 전기영동의 결과 DNA의 band가 다른 시료들보다 더 낮은 위치에서 관찰 되었다. 나머지 시료 중 H5과 H6 시료는 앞서 언급된 시 료들의 중간 정도되는 품질을 가졌으며 band의 위치가 H1, H2 시료 보다 조금 낮은 부위에서 발견되었다. Fig.
1b는 PstI 제한 효소가 처리된 DNA의 agarose gel 전기영 동 결과를 보여주고 있으며 Fig. 1a에서 관찰된 결과와 비 슷한 경향성을 보여주고 있다. 이와 같이, 본 연구에서 사 용된 DNA 시료는 다른 선행연구와 비슷하게 높은 수준으 로 degradation 이 진행된 DNA(H3, H4) 에서부터 거의 온전한 수준의 DNA(H1, H2) 까지를 포괄적으로 포함하고 있었다(Hughes-Stamm et al., 2011).
2. Genotype-by-sequencing을 통한 단편서열 생산
GBS를 통해 생성된 단편서열(sequencing read)을 대상 으로 index 서열에 따라 de-multiplexing을 진행한 결과, 각 시료당 생성된 단편서열의 수를 Table 1에 정리하였다.
시료 H3, H4은 DNA degradation이 높은 수준으로 진행 되어 생성된 단편서열의 수가 다른 시료들에 비해 10배 이 상 적었다. 시료 H5, H6번은 DNA의 평균길이와 agarose
Fig. 1. Agarose gel electrophoresis of 6 Hanwoo genomes. a) Agarose gel electrophoresis of 6 Hanwoo genomes with differing DNA quality. The ladders located in the left and right ends are the size marker. b) Agarose gel electrophoresis after treatment of PstI restriction enzyme.
[Provider:earticle] Download by IP 118.70.52.165 at Monday, December 20, 2021 7:51 PM
www.earticle.net
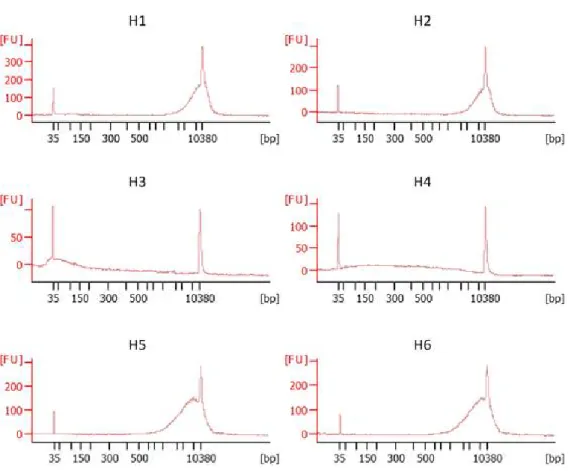
Fig. 2. Distribution of DNA size for each sample. Quantification of DNA using High Sensitivity DNA Assay is shown. The X-axis represents DNA size(bp) while the Y-axis shows the quantity of the DNA corresponding to each DNA size measured in low signal intensity(FU).
gel 전기영동 결과 모두에서 DNA degradation이 진행된 것을 확인했음에도 불구하고, 나머지 시료들과 DNA 단편 서열 생산량은 크게 차이가 나지 않았다. 결과적으로, 시료 H3와 H4에서만 확연하게 낮은 양의 단편서열이 생산된 것으로 보아, DNA degradation이 H3, H4와 같이 아주 높 은 수준으로 진행된 경우에 단편서열의 생산량이 영향을 받는다고 할 수 있다. 이는 DNA degradation에 의한 정보 손실을 보고한 선행연구와 비슷한 결과를 보여준다
(Graham et al., 2015). 그러나 H5, H6의 경우는 다른 해석 이 필요하다. GBS를 통하여 생산하는 DNA는 특정 DNA 서열을 대상으로 하는 제한 효소의 인식부위에 크게 영향 이 없었다고 볼 수 있으며, 이는 GBS 방법 특성상 절단된 단편 중 150-300bp의 작은 절편만을 대상으로 하는 것과 맞물려 나타난 결과로 해석된다. 따라서 H5, H6 시료의 DNA degradation정도가 GBS의 자료생산량에 영향을 끼 칠 확률이 낮을 것으로 예상된다.
Table 1. Summary of Genotype-by-sequencing
Sample ID Total reads Retained reads Average base quality Alignment rate (%) SNP calling
H1 1,572,363 1,218,206 32.1 94.32% 66,424
H2 1,790,321 1,402,712 32.1 94.35% 70,273
H3 126,940 119,710 30.7 0.40% 67
H4 169,392 112,309 31.5 22.78% 4,178
H5 2,054,740 1,490,033 32.0 86.92% 68,596
H6 2,418,574 1,722,220 32.0 90.98% 70,366
[Provider:earticle] Download by IP 118.70.52.165 at Monday, December 20, 2021 7:51 PM
www.earticle.net
3. 참조 유전체 정렬을 통한 SNP발굴
차세대 염기서열분석의 결과로 얻을 수 있는 DNA 서열 은 각 염기 별로 품질점수를 가진다. 품질점수가 낮을수록 해당 염기가 오류일 확률이 증가하며, 이러한 염기들은 re-sequencing 과정에 오류를 일으킬 수 있으므로 제거되 어야 한다. 이러한 DNA 염기들을 제거하기 위하여 본 연 구에서는 전처리 과정으로 Trimmomatic 프로그램을 사용 하였다. Trimmomatic 프로그램에 의해 제거된 단편서열의 개수는 Table 1에 요약되어 있다. 이 과정을 통하여 품질점 수가 낮은 염기를 제거한 후 추가적으로 길이가 75bp 보다 짧은 단편서열들이 제거되는데, 각 시료에서 평균적으로 약 70%의 단편서열이 확보되었으나, DNA 의 품질이 가장 낮은 H3에서는 ~95% 정도의 단편서열이 확보되었다. 이 는 GBS 과정에서 길이가 충분한 일부DNA 서열이 높은 비중으로 선택되어 sequencing 되었다는 것을 뜻한다. 한 편 H3 다음으로 degradation이 심한 H4 는 다른 시료에 비해 높은 비중의 단편서열이(33%) 제거되었으며, 이는 DNA degradation 에 영향을 받아 조각난 많은 수의 단편 서열이 sequencing되었기 때문이라고 예상된다.
품질이 낮은 단편서열을 제거한 후, 나머지 단편서열들 은 소의 참조 유전체에 Bowtie2 프로그램을 이용하여 정렬 되었다. 이때, 참조유전체에 정렬된 단편서열의 양은 약 90% 정도인 것을 확인하였다. 이는 한우의 유전체를 참조 유전체인 Bos Taurus UMD3.1에 정렬한 결과로서 한우 품 종특이적 서열을 고려한다면 적당한 수치라고 할 수 있다. 실제로 한우의 전장 유전체를 sequencing한 연구에서도 이와 비슷한 수치를 보였다(Choi et al., 2015). 반면, DNA degradation이 높은 수준으로 진행된 시료 H3와 H4은 각 각 0.4, 22.78%의 DNA만이 참조유전체에 정렬되었다. 이 렇게 낮은 수치의 alignment rate는 두 가지 원인에 의해서 발생할 수 있다. 첫 번째 이유는 DNA degradation 및 변 형에 의한 오류이다. DNA degradation및 변형에 영향을 받은 DNA 서열은 선행연구에서 관찰하였듯이 치환 (substitution), 삽입(insertion) 혹은 결실(deletion)이 생겨 참조 유전체의 염기서열과 유의한 차이를 보일 수 있고 (Friedberg et al., 2005, Brotherton et al., 2007), 이는 단편 서열이 정렬되는 것을 방해할 수 있다. 또 하나의 원인으 로는 각 시료 특이적인 서열이다. 한우의 전장 유전체 연 구에서 발견했듯이, 한우에서는 참조유전체로 쓰인 다른 외래 품종의 소에서는 발견되지 않는 지역이 존재한다(Lee et al., 2013). 이와 같이 참조유전체에서 존재하지 않고
H3, H4번 샘플에서만 존재하는 부분에 제한 효소의 인식 부위가 존재하거나, 변이에 의해 제한 효소의 인식부위가 만들어 진다면, 그러한 부분이 sequencing되어 참조유전체 에는 정렬되지 않을 수 있다.
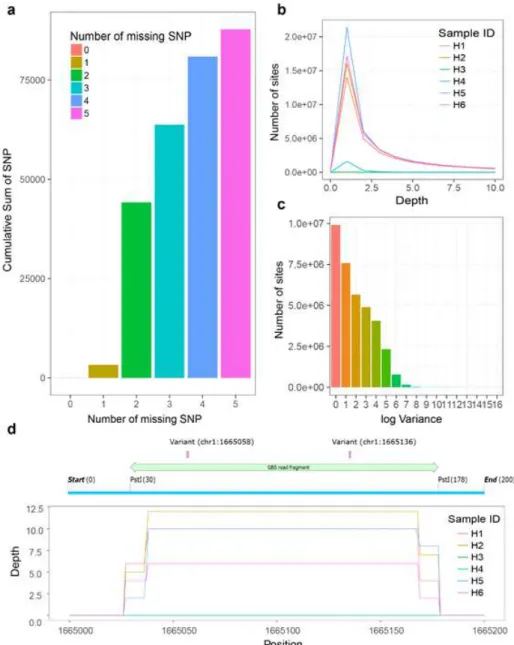
Alignment rate외에도 한우 유전체 정보를 참조 유전체 에 정렬하는 과정에 중요한 점은 sequencing된 단편서열 이 유전체의 동일한 부분에서 생성되었냐는 점이다. 이를 파악하기 위하여 유전체 각 좌표의 depth에 대한 분석을 수행한 결과가 Fig. 3b와 c에 나타나 있다. H3, H4를 제외 한 시료 별 depth의 분포는 Fig. 3b에 나타나 있듯이 서로 비슷한 것을 알 수 있다. 이는 전장유전체 전체를 보았을 때 평균적인 자료 생산량이 비슷한 수준이라는 것을 나타 낸다. 또한 각 좌표 별 depth의 분산은 같은 좌표에 쌓여 있는 depth가 비슷한 수준인가를 나타낸다. Fig. 3c에 나타 나 있는 histogram은 대부분의 좌표에서 각 시료 별 depth 가 비슷하다는 것을 나타낸다.
최종적으로 re-sequencing과정을 통해 발굴한 단일 염기 다형성(SNP) 은 유전적 지표로서 사용될 수 있다. 이러한 SNP 의 개수 또한 각 시료 별 확보된 단편서열의 개수 및 alignment rate를 포함한 앞서 언급된 수치들과 비슷한 경 향성을 보이고 있다. DNA 의 품질에 따라 발굴 할 수 있 는 SNP의 수가 Table 1에 요약되어 있다. Degradation이 높은 수준으로 진행된 H3에서 가장 적은 수의 SNP이 발 굴되었으며, 그 다음으로 DNA 품질이 낮은 H4 또한 두 번째로 가장 적은 수의 SNP이 발굴되었다. 이는 정렬된 단편서열의 수를 고려한다면 당연한 수치라고 할 수 있다. 이러한 경향성을 보여주기 위한 예로, Fig. 3d는 품질점수 가 높은 SNP 중 하나인(Chromosome 1, 1,665,058와 1,665,136)의 주변에서 관찰되는 각 시료의 depth를 보여주 고 있다. H3, H4 시료는 depth 0이 관찰되었고, 따라서 이 두 샘플에서는 유전자형을 유추할 수 없었다. 많은 수의 SNP에서 이러한 현상이 관찰되었으며, 이는 H3, H4의 SNP 수가 적은 것을 뒷받침 한다. 한편, 나머지 시료들에 서는 DNA 의 품질이 차이가 났음에도 불구하고, 6~70,000 개로 서로 비슷한 개수의 SNP을 발굴 할 수 있었다(Fig.
3a). 이러한 SNP 중, 34개 만이 모든 시료에서 관찰되었지 만, DNA 품질이 낮은 H3, H4시료를 제외한 경우에는
~40,000 개 정도의 SNP가 모든 샘플에서 발견되었다. 품질 이 가장 높았던 H1, H2 와 비교적 품질이 낮던 H5, H6번 샘플에서 발견된 SNP의 개수가 비슷하게 관찰되었던 것은 일정 수준의 degradation은 GBS 및 유전적 지표 발굴에 큰 영향을 미치지 않음을 뜻한다.
[Provider:earticle] Download by IP 118.70.52.165 at Monday, December 20, 2021 7:51 PM
www.earticle.net
Fig. 3. Re-sequencing analysis result. a) Number of SNP corresponding to the number of missing. b) Distribution of depth for each site. c) Histogram of log(variance) of depth for each site. d) The depth distribution near SNPs, including the ones located in chromosome 1, 1,665,058 and 1,665,136.
결론적으로, GBS를 통하여 sequencing한 단편서열은 DNA degradation에 의해 그 생산량이 영향을 받는다. 또 한 degradation의 정도는 생산된 단편서열에도 영향을 끼 칠 것으로 예상된다. 이는 단편서열 정렬의 정확성을 크게 감소시킬 수 있으며, 이를 통해 발굴 할 수 있는 SNP의 개 수도 크게 줄어들 수 있다. 하지만 이러한 현상은 DNA degradation이 높은 수준으로 일어났다는 전제하에 발생 하는 것으로 보이며, 본 연구는 일정 수준의 degradation (H5, H6정도의 수준)에 대해서는 GBS가 유전적 지표 발굴 에 강경한 방법이라는 점을 밝혔다.
Ⅳ. 요약
Genotyping-by-sequencing(GBS)은 저렴하면서도 효율 적인 유전적 표지 발굴 방법으로, 집단 분석을 통한 SNP(single nucleotide polymorphism) 발굴에 있어 사용 될 수 있다. 제한 효소를 이용하여 유전체의 일부분을 sequencing 하는 GBS의 특성상, DNA의 품질은 GBS 연구 의 후속 과정에 영향을 끼칠 수 있다. 그러나 DNA degradation 및 품질이 GBS 기반 유전적 지표 발굴에 있 어서 끼치는 영향에 대한 연구는 많이 이루어지지 않은 상
[Provider:earticle] Download by IP 118.70.52.165 at Monday, December 20, 2021 7:51 PM
www.earticle.net
황이다. 본 연구는 DNA degradation 정도가 다른 한우 집 단 GBS 자료를 이용하여 비교분석을 수행하였다.
Re-sequencing 파이프라인을 따라서 DNA degradation이 유전적 표지 발굴에 어떠한 영향을 끼치는지 조사하였으 며, 그 결과, DNA degradation이 일부 진행된 H5, H6 시 료에서는 degradation이 SNP의 양과 품질에 큰 영향을 끼 치지 않는다는 것을 확인하였고, 높은 수준으로 DNA degradation이 진행된 경우(H3, H4) 유전적 표지 발굴에 있어서 문제가 된다는 것을 확인하였다. 이는 GBS 연구가 일정 수준의 degradation에 대해서는 강경한 방법이라는 점을 뒷받침 한다.
사사
본 논문은 농촌진흥청 차세대 바이오그린21사업(과제번호:
PJ01134905)의 지원에 의해 이루어진 것임.
Ⅴ. 참고문헌
1. Bolger, A. M., Lohse, M. and Usadel, B. 2014.
Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114-2120.
2. Brotherton, P., Endicott, P., Sanchez, J. J., Beaumont, M., Barnett, R., Austin, J. and Cooper, A. 2007. Novel high-resolution characterization of ancient DNA reveals C> U-type base modification events as the sole cause of post mortem miscoding lesions. Nucleic Acids Res. 35(17):5717-5728.
3. Bruskov, V. I., Malakhova, L. V., Masalimov, Z. K.
and Chernikov, A. V. 2002. Heat-induced formation of reactive oxygen species and 8-oxoguanine, a biomarker of damage to DNA. Nucleic Acids Res.
30(6):1354-1363.
4. Cadet, J., Sage, E. and Douki, T. 2005. Ultraviolet radiation-mediated damage to cellular DNA. Mutat.
Res. 571(1):3-17.
5. Choi, J. W., Choi, B. H., Lee, S. H., Lee, S. S., Kim, H.
C., Yu, D., Chung, W. H., Lee, K. T., Chai, H. H. and Cho, Y. M. 2015. Whole-genome resequencing analysis
of Hanwoo and Yanbian cattle to identify genome-wide SNPs and signatures of selection. Mol.
Cells 38(5):466.
6. De Donato, M., Peters, S. O., Mitchell, S. E., Hussain, T. and Imumorin, I. G. 2013. Genotyping-by- sequencing (GBS): a novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PLoS One 8(5):e62137.
7. Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S. and Mitchell, S. E. 2011.
A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6(5):e19379.
8. Friedberg, E. C., Walker, G. C., Siede, W. and Wood, R. D. 2005. DNA Repair and Mutagenesis, American Society for Microbiology Press. USA. pp. 258-259.
9. Graham, C. F., Glenn, T. C., McArthur, A. G., Boreham, D. R., Kieran, T., Lance, S., Manzon, R. G., Martino, J. A., Pierson, T. and Rogers, S. M. 2015.
Impacts of degraded DNA on restriction enzyme associated DNA sequencing (RADSeq). Mol. Ecol.
Resour. 15(6):1304-1315.
10. He, J., Zhao, X., Laroche, A., Lu, Z. X., Liu, H. and Li, Z. 2014. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 5:484.
11. Herten, K., Hestand, M. S., Vermeesch, J. R. and Van Houdt, J. K. 2015. GBSX: a toolkit for experimental design and demultiplexing genotyping by sequencing experiments. BMC Bioinformatics 16(1):73.
12. Hofreiter, M., Jaenicke, V., Serre, D., Haeseler, A. v.
and Pääbo, S. 2001. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res.
29(23):4793-4799.
13. Hughes-Stamm, S. R., Ashton, K. J. and van Daal, A.
2011. Assessment of DNA degradation and the genotyping success of highly degraded samples. Int.
J. Legal Med. 125(3):341-348.
14. Langmead, B. and Salzberg, S. L. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9(4):357-359.
[Provider:earticle] Download by IP 118.70.52.165 at Monday, December 20, 2021 7:51 PM
www.earticle.net
15. Lee, K. T., Chung, W. H., Lee, S. Y., Choi, J. W., Kim, J., Lim, D., Lee, S., Jang, G. W., Kim, B. and Choy, Y. H. 2013. Whole-genome resequencing of Hanwoo (Korean cattle) and insight into regions of homozygosity. BMC Genomics 14(1):519.
16. Lindahl, T. 1993. Instability and decay of the primary structure of DNA. Nature 362(6422):709-715.
17. Lindahl, T. and Nyberg, B. 1972. Rate of depurination of native deoxyribonucleic acid. Biochemistry 11(19):3610-3618.
18. McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S. and Daly, M. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data.
Genome Res. 20(9):1297-1303.
(Received 07 November 2017, Revised 13 December 2017, Accepted 14 December 2017)
[Provider:earticle] Download by IP 118.70.52.165 at Monday, December 20, 2021 7:51 PM