DOI 10.17480/psk.2018.62.3.179
신약 후보물질 발굴을 위한 비임상포뮬레이션 전략
강원호*·채정우*·윤휘열#
*충남대학교 약학대학
(Received January 23, 2018; Revised June 11, 2018; Accepted June 18, 2018)
Preclinical formulation strategies for discovering new drug candidates
Won-ho Kang*, Jung-woo Chae* and Hwi-yeol Yun#
*College of Pharmacy, Chungnam National University, 34134, Daehak-ro 99, Yuseonggu, Daejeon, Republic of Korea
Abstract — To determine a rational preclinical formulation strategy for preclinical research, scientists should consider with their own work-flow and physicochemical properties acquired by each discovery stage. In general, physicochemical prop- erties such as ionization states and kinetic solubility could be obtained in the early discovery stage. However, more detailed properties such as equilibrium solubility, pKa, pH-solubility and cLog P/D could be confirmed at the late discovery stage.
For these reasons, it was hard to decide which kinds of formulation were most suitable for candidates. Therefore, the pri- mary objective of this review was to figure out preclinical formulation strategy based on drug discovery stage, and propose case-study with real case for application. Preclinical formulation strategy of previously approved drug that has similar char- acteristics usually was used in early drug discovery stage. Especially if information of drug characteristics was limited, researchers were able to consider that it was alternative methods to make suspension with surfactant and cyclodextrin-drug complexation method. For late discovery stage, there are several ways to consider the characteristic of various phys- icochemical properties such as pH-adjustment, solubilization (using by co-solvent, surfactant) and combination of pH-adjust- ment and solubilization. As drug discovery work-flow are different among pharmaceutical companies, making preclinical formulation strategy from early stage of drug discovery is important for increment of success ratio of new drug devel- opment. This review could guide researchers which preclinical formulation is appropriate by suggesting rational strategy.
Keywords Preclinical formulation strategy, physicochemical properties, solubilization, suspension
서 론
신규화합물을 이용한 비임상 시험 진행 시 발생하는 여러 문 제들 중, ‘어떤 부형제(vehicle)를 이용하여 어떠한 성상으로 약 물을 투여할 것인가?’ 하는 문제는 약물의 흡수율을 결정짓는 중 요한 부분 중 하나이다.1,2,3) 추가적으로 최근, 신약개발 시 분자 량이 커지고 지용성이 높은 난용성 물질들의 개발이 많아지는 추 세이고,4,5)중개연구의 활성화로 개발 초기 단계에서 치료 용량
탐색을 위해 고용량 투여가 필요한 상황이다. 이때 부형제로 인 한 노출 영향(formulation effect)을 줄이기 위하여 균질한 현탁 화, 용액화 등을 위한 부형제 선택 및 투여물질 제조방법에 대한 고민이 필요하다.6,7,8)그리고 약물 개발 초기 단계에서 약물의 정확한 절대생체이용률 파악 및 약물의 분포, 대사, 배설 과정 (disposition)을 이해하기 위하여 정맥 투여 또한 필요하기 때문 에 약물을 가용화 시킬 수 있는 전략이 필수적이다. 이 과정에서 약물의 가용화, 균질화 뿐만 아니라 각종 첨가제들이 일으킬 수 있는 독성 증상 또한 고려하지 않을 수 없다.6,7,9) 그 동안 전형 적인 신약개발의 과정의 초기발견단계(early discovery stage)에 서는 비임상 연구자들에게 시험 물질을 포함한 물리화학적 정보 가 전달되지 않는 실정이다.6) 따라서 비임상 연구자들은 단순히 답습적으로 사용하던 부형제들을 과학적 근거 없이 경험적으로 판단하여 사용해 왔다. 이러한 결과, 상대적으로 많은 정보를 얻
#
Corresponding Author Hwi-yeol Yun
College of Pharmacy, Chungnam National University, Daejeon 34134, Republic of Korea
Tel.: 042-821-5941 Fax.: 042-823-6781 E-mail: [email protected]
Short Report
종설각종첨가제 등을 사용해야 하는지 알지 못하였으며, 이로 인해 초 기발견단계에서 사용하던 부형제를 약동학(PK, pharmacokinetics) 또는 약력학(PD, pharmacodynamics) 프로파일 차이를 우려하여 후기발견단계에서 그대로 사용해오고 있다.
이 논문의 목적은 신약개발의 성공적인 비임상 연구 수행을 위해 새롭게 합성된 유기화합물의 물리화학적 정보가 절대적으 로 부족한 초기발견단계와 어느 정도 물리화학적 정보를 획득하 게 되는 후기발견단계에서 각각 어떠한 요소들을 근거로 최적의 비임상포뮬레이션(preclinical formulation) 전략을 확립할 수 있 을 지에 대해 제안하고자 한다.
본 론
일반적인 신약 발견 과정
일반적인 신약발견과정(drug discovery process)은 먼저 개발 된 신약들의 모방(mimic) 화합물들을 유기합성하여 타겟 단백질 또는 세포에서의 활성을 평가하고 병태 동물모델에서 실제 약효 를 확인한다. 물론 최근의 트렌드는 시험관 내 활성(in-vitro activity)평가와 생체 내 효력(in-vivo efficacy)평가 사이에 시험관 내 흡수, 분포, 대사, 배설(in-vitro Absorption, Distribution, Metabolism and Elimination) 평가 및 생체 내 약동학 평가를 통 하여 후보물질들의 약물유사성(drug-likeness)을 판단하고 생체 내 효력 평가를 수행하는 방식으로 신약발견 work-flow의 효율 성 개선을 시도하는 걸로 변화하고 있지만,10,11,12)여전히 많은 회사에서는 초기발견단계의 핵심목표로 신약후보물질의 약물잠 재력(potency)과 생체내 효력(efficacy)확인에 초점을 두고 있기 에 시험관 내 약동학 평가를 신약개발의 후기발견단계(late discovery stage)에서 수행하는 경우가 여전히 많다6)(Fig. 1). 선 도물질들이 발굴되고 최적화 단계를 진행하게 되는 후기발견단 계에서는 2차 약효(secondary efficacy) 평가, 흡수, 분포, 대사, 배설특성 규명(ADME characterization) 및 단기 독성(short term toxicity)등에 대한 평가들이 이루어지게 되고 이 단계에서 핵심은 여러 선도물질들의 흡수, 분포, 대사, 배설특성, 독성증상 규명 및 약효에 대한 확증이 이루어진다.10,11,12) (Fig. 1)
신약후보물질의 물리화학적 요소들이 흡수, 분포, 대사, 배설에 미치는 영향
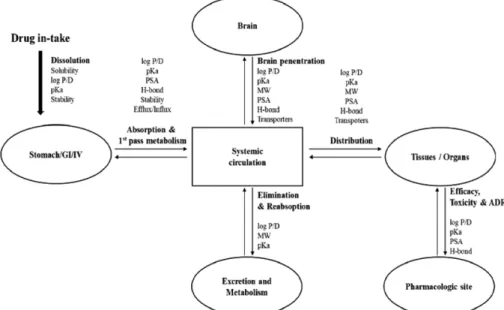
유기 화합물의 물리화학적 요소들은 제제 연구자(formulation scientist)들에게 대단히 중요한 정보이지만 단지 프리포뮬레이션 (preformulation) 및 포뮬레이션(formulation)을 위한 정보만이 아 니라 생체 내에서 일어나는 모든 현상에 이 물리화학적 요소가 관여하고 있기 때문에 비임상 및 임상 연구자들에게도 매우 중
요한 정보라고 할 수 있다. 각 후보물질들의 물리화학적 특성이 약물의 흡수, 분포, 대사, 배설에 미치는 영향은 Fig. 2와 같이 정 리할 수 있다.4,13,14,15)
신약발견 단계 별 물리화학적 요소
일반적으로 초기발견단계에서는 화합물의 이온화 상태(ionization state), kinetic 용해도(kinetic solubility)등의 정보가 주어지고 투 여 용량은 통상적으로 10 mg/kg이상으로 하게 된다.(Fig. 3) 이 단계에서 비임상 연구자들이 선택할 수 있는 부형제 선택 전략 은 거의 없다. 왜냐하면 이온화 상태를 알더라도 산해리상수(pKa) 와 pH 별 용해도를 알 수 없기 때문에 이 요소를 이용하여 부 형제 선택을 할 수 없게 된다. 그리고 kinetic 용해도의 경우 Dimethyl Sulfoxide (DMSO) 와 같은 양극성 유기 용매에 활성 원료(Active Pharmaceutical Ingredients, API) 를 충분히 녹인 후 aqueous buffer에 첨가하여 용해도를 측정하므로 일반적으로 equilibrium 용해도에 비해 높게 측정된다.13)그리고 최근 부형 제 선택에 있어 DMSO와 같은 유기 용매는 중대한 독성증상 (vesiculation, a sensation of burning, local allergic, dryness of skin) 및 초기 대사연구(early metabolic study)에서 DMSO로 인한 다양한 변수(막 투과도 및 담즙 분비 촉진 등)가 결과에 영 향을 줄 수 있어 부형제로 선택하는 것을 지양하는 추세이다.16) 후기발견단계에서는 초기발견단계에 비해 상대적으로 많은 물 리화학적 정보들을 얻게 된다(ionization state, kinetic solubility 외에도 equilibrium solubility, pKa, pH-solubility, cLog P/D 등) (Fig. 3.) 그리고 초기발견단계에서의 약동학, 약력학 데이터를 근
Fig. 1 − Kang et al., General work flow in drug discovery stage
(Cytotox : cytotoxicity, P.S. : Plasma stability, M.S. :
Metabolic stability, CYP inh. : CYP inhibition, hERG inh :
hERG inhibition, PPB : Plasma protein binding).
거로 약효가 발현될 것으로 예상되는 용량과 공비 또는 공차를 이용해 2개 이상 용량을 추가하여 용량에 따른 약동학, 약력학 적 변화를 측정하게 된다.
신약발견 단계별 기존 비임상포뮬레이션 전략
초기발견단계에서는 앞 단락에서 기술한 바와 같이 부형제 선 택에 대한 정보가 거의 없는 상태에서 각 연구팀 별로 상황에 맞 는 전략을 크게 3가지 정도로 설정하여 수행하고 있다.
우선 첫번째로 모방 화합물을 만들고 있다면, 동일계열 선행 물질의 비임상 또는 초기 임상 시험에 사용한 부형제를 사용하 는 방법이다. 정맥투여가 가능한 부형제가 더 선호된다. 하지만 이 전략은 모방 화합물을 합성할 때 타겟 단백질과의 친화도 향 상 등을 위하여 이온화 가능 그룹(ionizable functional group)을 이온화 불가능 그룹(non-ionizable functional group)으로 변경하 여 합성할 때가 있어 모든 케이스에 적용할 수 있는 전략은 아 니라고 할 수 있다.
두번째 전략은 물리화학적 요소들을 무시하고 현탁화 시키는 방법이다. 대부분의 합성화합물들은 이온형이고 약염기성 화합 물이므로 산성 환경인 위장에서의 약물의 용해도가 높다고 가정 하고 현탁화 시켜 투여하는 방법이다. 그리고 물에서의 뭉침(clumpy) 현상 등을 방지하기 위해서 약간의 첨가제(예: 가용화제)를 첨가하 여 최대한 균질한 현탁제를 제조하여 투여하는 것이 더 선호된 다. 그러나 이 전략은 심각한 난용성 화합물의 경우, 저 용량에 서도 정맥투여를 위한 용액을 제조할 수 없는 방법이다. 따라서 이러한 난용성 화합물의 정맥투여를 위해서는 또 다른 부형제를 고려할 수 밖에 없다.
마지막 전략은 Cyclodextrin계열의 부형제를 이용한 용액화 전 략이다. 주로 Hydroxypropyl beta cyclodextrin (HP-β-CD)를 이 용하거나 HP-β-CD의 신독성 문제를 개선한 Sulfobutyl ether
Fig. 2 − Kang et al., Schematic presentation of the relationship and interplay of physicochemical properties with in vivo drug pharmacokinetic/
pharmacodynamic and exposure
4)(Modified from Jiliang et al., GI : Gastrointestinal tract, IV : Intravenous, log P : Partition coefficient in a lipophilic and aqueous phase, log D : Distribution coefficient in a lipophilic and aqueous phase, pKa : Acid dissociation constant, PSA : Polar surface area, MW : Molecular weight, H-bond : Hydrogen bond, ADR : Adverse drug reaction).
Fig. 3 − Kang et al., Obtainable Physicochemical properties in early
and late discovery stage.
물분자의 안정성 향상에 도움이 된다. 그러나 cyclodextrin과 약 물이 형성한 포접으로부터 약물분자가 해리되어 나오지 않으면 약물이 위장관에서 투과될 수 없어서 흡수의 측면에서 오히려 불 리한 요인이 되기도 한다.17) (Fig. 4)
초기발견단계와는 달리, 후기발견단계에서는 확보한 많은 물 리화학적 요소들(ionization state, kinetic solubility, equilibrium solubility, pKa, pH-solubility, cLog P/D 등)을 바탕으로 다양한 전략들을 동원해 볼 수 있다. 첫번째 이온화상태(ionization state), 산해리상수(pKa), equilibrium 용해도(equilibrium solubility)를 고려한 pH 조정(pH-adjustment)방법을 생각해 볼 수 있다. 즉 산성약물은 염기성완충액에 염기성약물은 산성완충액에 넣어 용 해시켜보는 방법이다. 이 때 사용 되는 완충액 종류는 산성완충 액의 경우 10 mM citrate buffer(pH 3~4.5)와 10 mM Acetate buffer(pH 4.5~6)등이 있으며 염기성완충액의 경우 10 mM phosphate buffer(pH 6~8.5)와 10 mM carbonate buffer(pH 8.5~9)등이 있다. 하지만 pH 조정 방법은 높은 용해도를 나타내 는 pH범위에서의 용해도가 실험하고자 하는 용량을 커버하지 못 할 수 있으며 산성약물의 경우에는 경구투여 시 위액의 pH가 매 우 낮기 때문에 염기성완충액에 용액화 되었더라도 경구투여 후 위장에서부터 석출현상을 피할 수 없다. 18, 19) 두번째 전략은 약 물의 지용해도(lipophilicity; Log P/D)를 고려하여 lipid based formulation을 만들어 보는 방법이다. 즉 지용성이 높은 화합물 들을 지용성(lipidic) 첨가제를 이용해 용해시켜보는 방법이다. 이 때 사용되는 지용성 첨가제들에는 Soybean Oil(50-100%),
제들을 이용하여도 실험하고자 하는 용량을 커버하지 못할 수 있 다. 그리고 이러한 lipid based formulation의 경우에는 위장액 내 에서 소포(vesicle)또는 미셀(micelle)을 형성하며 지방의 소화과 정을 거치게 되는데 이 때 약물 분자가 소포 또는 미셀로부터 빠 져 나오지 못한다면 투과되지 못하여 흡수율이 낮아질 수 있다.
그리고 장기간 투여 시 발생할 수 있는 이상증상(diarrhea 등)또 한 고려하지 않을 수 없다.19,20,21)세번째 전략은 equilibrium 용 해도를 고려하여 용해보조제(solubilizer)를 활용한 용액화 전략 (Solubilization)이다. 즉 용해도를 높여 과포화 상태의 용액으로 만들어 보는 방법이다. 이때 사용되는 용해보조제(solubilizer)로는 Tween80(5~10%), Cremophor EL(5~10%), Pluronic F68 (20~60%), Solutol HS-15(10~50%), Vitamin E-TPGS(10~50%), Gelucire 44/14(20~50%), Labrasol(40~60%)등이 있다.9,18,19)그 러나 앞의 경우들과 마찬가지로 이러한 용해보조제(solubilizer)들 을 이용하여도 실험하고자 하는 용량을 커버할 수 있을 만큼의 용해도가 향상되지 못할 수도 있으며 또 이러한 첨가제(surfactant) 들이 일으킬 수 있는 여러가지 독성증상(diarrhea, irritation 등) 에 대해서 고려하지 않을 수 없다. 마지막으로 화합물의 활성에 는 영향을 주지 않으면서 수 용해도(aqueous solubility)를 향상 시킬 수 있도록 염(salt)를 도입하거나 수화물(hydrate)을 만드는 방법이 있을 수 있다.22)이 방법은 비임상포뮬레이션 전략이기 보다는 화학적 변경을 통한 수용해도 향상이므로 본 논문에서는 더 이상 기재하지 않기로 한다. 그 밖에 앞서 소개한 pH조정과 용액화 전략을 함께 사용하는 방법이 있을 수 있다.(Fig. 4)
Fig. 4 − Kang et al., Preclinical Formulation strategies in early and late discovery stage.
후기발견단계(late discovery stage)에서는 약물의 생체이용률 파악 및 정확한 약물의 분포, 대사, 배설 양상(disposition)을 이 해하기 위해 정맥 및 임상적용경로 투여를 진행하게 되는 단계 로 위에 소개된 용액화(solubilization) 방법들을 이용하여 약물을 투여함으로써 여러 가지 우려사항을 제거할 수 있다. 하지만 심 각한 난용성 약물의 경우 정맥투여시에는 용액(저 용량)으로 경 구투여 시에는 현탁액(효력 용량)으로 투여 하게 되는데 이때 산 출된 경구 생체이용률은 위장관내에서의 부형제로 인한 노출 영 향을 고려하여 생각해야 한다. 약물이 위장관내에서 용해도의 이 유로 흡수에 한계에 부딪히게 되면 비선형 약물동태의 양상을 보 이게 되므로 용량 별 PK 결과를 바탕으로 위장관내에서 용해도 에 의한 영향이 있는지 파악할 수 있다. 이러한 경우에는 임상시 험전 가용화연구와 같은 제제학적 연구를 통해 용해도 및 용출 개선을 시도하는 것이 좋다.
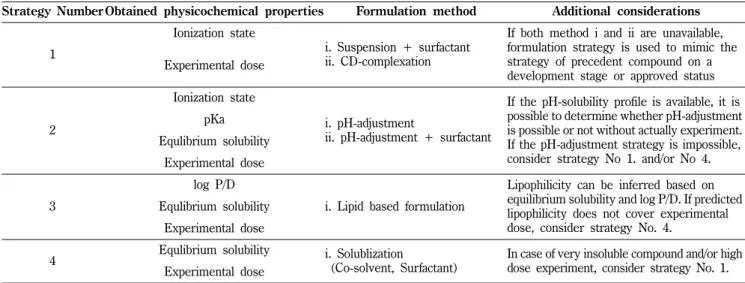
물리화학적 요소에 따른 비임상포뮬레이션 전략 제시
기존 신약 개발 단계별 비임상포뮬레이션 전략은 모두 완벽하 지는 않으며, 회사마다 모두 신약 발견 work-flow와 프리포뮬레 이션 연구가 시작되는 시기가 다를 수 있다. 따라서 기존에 사용 해 오던 신약 발견 단계별 비임상포뮬레이션 전략을 보완하기 위 해 획득한 물리화학적 정보에 따른 전략을 제안해 보았다.(Table 1) 우선 주어진 정보가 Ionization state와 실험 용량만 알고 있 다면 고려할 수 있는 비임상포뮬레이션 전략은 현탁액(+가용화 제), Cyclodextrin-complexation 그리고 개발 단계에 있는 선행 물질들의 비임상포뮬레이션 전략을 모방하여 사용해볼 수 있을 것이다. 두번째로 이온화상태(Ionization state), 산해리상수(pKa), equilibrium 용해도(equilibrium solubility)와 실험 용량을 알고 있다면 pH-조정(+가용화제) 전략을 선택해 볼 수 있다. 셋째 log P/D, equilibrium 용해도(equilibrium solubility)와 실험 용량을
알고 있다면 lipid based formulation 전략을 선택해 볼 수 있다.
마지막으로 equilibrium 용해도(equilibrium solubility)와 실험 용 량만을 알고 있다면 용액화(co-solvent and/or surfactant) 전략 을 선택해 볼 수 있다.
적용 예시(화합물 X)
분자량 342.2 g/mol의 약염기성 화합물 X의 정보가 Table 2와 같다고 할 때, 화합물 X의 비임상포뮬레이션 전략에 대해 논의 해 보기로 한다.
우선 이온화상태(ionization state), 산해리상수(pKa), equilibrium 용해도(equilibrium solubility)를 알고 있으므로 pH-조정 방법에 대 해서 생각해 본다. 산해리상수가 8.9이고 equilibrium 용해도가 0.074 mg/ml인 약염기성 화합물은 Henderson-Hasselbalch 방정 식에 따르면 약 pH 7이하의 구간에서는 모두 0.074 mg/ml의 용 해도를 유지할 것이므로 pH-조정 방법으로 이 화합물을 용해시 키기에는 어려움이 있을 것으로 판단된다. 두번째 방법은 Log P 를 알고 있으므로 lipid based formulation을 생각해 볼 수 있다.
그러나 Log P가 약 2.88인 화합물의 지용해도는 최대 약 0.2 mg/
Table I − Preclinical formulation strategies according to known physicochemical properties
Strategy NumberObtained physicochemical properties Formulation method Additional considerations
1
Ionization state
i. Suspension + surfactant ii. CD-complexation
If both method i and ii are unavailable, formulation strategy is used to mimic the strategy of precedent compound on a development stage or approved status Experimental dose
2
Ionization state
i. pH-adjustment
ii. pH-adjustment + surfactant
If the pH-solubility profile is available, it is possible to determine whether pH-adjustment is possible or not without actually experiment.
If the pH-adjustment strategy is impossible, consider strategy No 1. and/or No 4.
pKa Equlibrium solubility
Experimental dose
3
log P/D
i. Lipid based formulation
Lipophilicity can be inferred based on equilibrium solubility and log P/D. If predicted lipophilicity does not cover experimental dose, consider strategy No. 4.
Equlibrium solubility Experimental dose
4 Equlibrium solubility i. Solublization
(Co-solvent, Surfactant)
In case of very insoluble compound and/or high dose experiment, consider strategy No. 1.
Experimental dose
Table II − Physicochemical properties of compound X
Information Unit Value
Molecular weight g/mol 342.2
Ionization state - Weak basic
pKa - 8.9
Log P - 2.88
Equilibrium solubility(water) mg/ml 0.074
Experimental dose mg/Kg 20
Dosing volume ml/Kg 5
Experimental concentration mg/ml 4
을 알 수 있다. 용액화를 위하여 남은 방법은 co-solvent 및 co- solvent에 가용화제 첨가를 통한 용액화 방법이다. 실험에 사용 할 화합물의 농도와 실제 수용해도 사이에는 약 54배 정도의 차 이가 있으므로 이 정도의 차이를 극복할 수 있는 가용화제를 동원 해야 하며 polypropylene glycol(30~60%) 또는 polyethylene glycol400(40~100%)과 물을 혼합한 co-solvent에 polysorbate80 (10% 이하) 또는 cremorphor EL(10% 이하)과 같은 가용화제를 첨 가하여 과포화상태의 용액을 조제할 수 있을 것이다.
결론 및 고찰
비임상 시험 수행 시 비임상포뮬레이션은 신약후보물질 발굴 을 위한 가장 중요한 요인 중 하나이다. 합리적인 비임상포뮬레 이션 전략을 결정하기 위해서 고려해야하는 부분으로는 비임상 평가 플로우(work-flow)와 각 평가 단계별로 확보되는 물리화학 적 요소들이다. 대부분 국내 제약사들의 비임상 평가 플로우는 후보물질들의 약물잠재력(potency)과 생체내 효력(efficacy)에 대 해 평가해 보는 초기발견단계(early discovery stage)와, 흡수, 분 포, 대사, 배설 및 독성에 대한 규명 그리고 약효에 대한 확증을 위해 여러 평가가 수행되는 후기발견단계(late discovery stage) 로 나누어 볼 수 있다. 이때 초기발견단계(early discovery stage) 에서 확보될 수 있는 물리화학적 요소들은 이온화 상태(ionization state), kinetic 용해도(kinetic solubility)등이 있으며 후기발견단 계(late discovery stage)에서 확보될 수 있는 물리화학적 요소들 은 이온화 상태, kinetic 용해도 외에도 equilibrium 용해도 (equilibrium solubility), 산해리상수(pKa), pH별 용해도, cLog P/
D 등이 있다. 각 단계별로 확보된 물리화학적 요소에 따라 비임 상포뮬레이션 전략을 생각해 볼 수 있다. 우선 초기발견단계(early discovery stage)에서는 기 허가된 동일 계열 물질의 formulation 전략을 따라가거나 가용화제가 일부 포함된 현탁제로 만드는 방 법 그리고 cyclodextrin류를 이용한 complexation방법이 있을 수 있다. 초기발견단계(early discovery stage)에서는 확보된 다양한 물리화학적 요소들의 특성을 감안하여 pH-조정 방법, lipid-base formulation을 만드는 방법, co-solvent 및 surfactant를 이용하여 과포화 용액으로 만드는 방법 그리고 pH-조정과 과포화 용액화 방법을 조합하여 용액화하는 방법 등이 있을 수 있다. 그러나 신 약발견 work-flow와 프리포뮬레이션 연구가 시작되는 시기는 회 사 마다 그리고 프로젝트의 특성에 따라 모두 다를 수 있어 빅 파마들의 프리포뮬레이션 팀에서는 신약후보물질들의 물리화학 적 특성을 고려한(discovery formulation decision tree)를 만들 어서 비임상포뮬레이션에 활용하고 있다.1,18)따라서 본 연구팀 은 신약발견 단계(초기 또는 후기)에 관계없이 확보한 물리화학
택하는데 있어 매우 결정적인 데이터가 된다. 이때 각 물질들의 물리화학적 요소를 반영한 합리적인 비임상포뮬레이션을 선정하 여 동물실험을 수행한다면 흡수 부분에 있어서 큰 우려사항 하 나를 제거할 수 있게 될 것이다. 더불어 초기발견단계에서부터 후보물질의 물리화학적 정보를 빠르게 파악하고 그 정보를 비임 상포뮬레이션 선정에 활용하여 후보물질에 대한 실제적 포뮬레 이션 경험들이 축적되게 된다면 추후 이를 초기 임상 포뮬레이 션에도 연결할 수 있을 것이다.3,23)또한 빠르게 파악된 물리화 학적 특성들을 정량적 구조-활성 상관관계(QSAR)와 생리학 기 반 약물동태 모델링(PBPK modeling)등에 활용할 수 있다면24) 신약후보물질 발굴을 가속화 할 수도 있을 것 이다.
감사의 말씀
이 연구는 충남대학교 학술연구비에 의해 지원되었으며, 이에 감사를 드립니다.
References
1) Suma, G., Amr, N. and Alan, G. W. : Development and application of a high-throughput formulation screening strategy for oral administration in drug discovery. Future Med.
Chem. 2, 1391 (2010).
2) Roy, H., Allen, T., Stephen, B. and Thomas, E. P. : Discovering and Developing Molecules with Optimal Drug-Like Properties.
Springer. New York(2015).
3) Margaret, S. L., Shobha, B., Mehran, Y. and John, M. : Commentary: Why Pharmaceutical Scientists in Early Drug Discovery Are Critical for Influencing the Design and Selection of Optimal Drug Candidates. AAPS PharmSciTech.
19, 1(2017).
4) Jianling, W. and Suzanne, S. : Recent advances in physicochemical and ADMET profiling in drug discovery.
CHEMISTRY & BIODIVERSITY. 6, 1887(2009).
5) Christel, A. S. B. and Mehran, Y. : Lipophilicity in Drug Development: Too Much or Not enough?. The AAPS Journal.
18, 1095(2016).
6) Seshadri, N. : Preclinical formulations for discovery and toxicology: physicochemical challenges. Expert Opin. Drug Metab. Toxicol. 2, 715(2006).
7) Sanket, M. S., Ankitkumar, S. J., Ritu, K., Mangal, S. N. and Maneesh, J. N. : Preclinical Formulations: Insight, Strategies, and Practical Considerations. AAPS PharmSciTech. 15, 1307(2014).
8) Paul, D. L. and Brian, S. : The influence of drug-like concepts on decision-making in medicinal chemistry. Nature Review Drug Discovery. 6, 881(2007).
9) Shayne, C. G., Nicolas, A., Bart, S., Heide, R. and Crystal, D.
C. : Nonclinical vehicle use in studies by multiple routes in multiple species. International Journal of toxicology. 25, 499(2006).
10) Ashutosh, K., Jennifer, R., Cindy, P., April, B., Andrew, C., Tao, S. and Mehran, F. M. : Proposing advancement criteria for efficient DMPK triage of new chemical entities. Future Med.
Chem. 6, 131(2014).
11) Mehran, F. M., Yang, T., Zhihong, O., Samantha, J. R., Maria, B., Prasoon, C., Julius, A. and Ashutosh, K. : A proposed screening paradigm for discovery of covalent inhibitor drugs.
Drug Metabolism Letter. 8, 19(2014).
12) Anna-Karin, S. S., Juliette, J., Johan, B., Pawel, B., Anna, B. E., Yin, H., Carrie, T., Anders, L., Olof, G., Tjerk, B., Sveinn, B., Sanja, J., Jenny, J., Margareta, B. and Janet, H. : Optimizing DMPK Properties: Experiences from Big Pharma DMPK Department. Current Drug Metabolism. 17, 253(2016).
13) Edward, H. K., Li, D. : Drug-like properties: Concepts, Structure design and method, Elsevier Inc. San Diego(2008).
14) Siamak, C. K., Harvey, W. and Cornelis, E. C. A. H. : Drug metabolism and pharmacokinetics quick guide. Springer, New York(2011).
15) John, A. A. and Sonia, L. P. : The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 7, 909(2012).
16) Patricia, V. T., Cynthia, P., Mary, A. V. and Thea, B. : Administration of substances to laboratory animals: Equipment considerations, vehicle selection, and solute preparation. Journal
of the American for Laboratory Animal Science. 50, 614(2011).
17) Thorsteinn, L., Maria, D. M. O., Carmen, A. L. and Angel, C. : Pharmacokinetics of cyclodextrins and drugs after oral and parenteral administration of drug/cyclodextrin complexes.
Journal of Pharmacy And Pharmacology. 68, 544(2016).
18) Yung-Chi, L., Philip, D. Z. and Brian, S. : An intravenous formulation decision tree for discovery compound formulation development. International Journal of Pharmaceutics. 253, 111(2003).
19) Ping, L. and Luwei, Z. : Developing early formulations: Practice and perspective. International Journal of Pharmaceutics. 341, 1(2007).
20) Sandeep, K., Mohanvarma, M. and Veerabhadhraswamy, P. : Oral lipid-based drug delivery system – an overview. Acta Pharmaceutica Sinica B. 3, 361(2003).
21) Orlagh, M. F., Matthew, F. C., Claire, L. M., Natalie, L. T., Hywel, D. W., Colin, W. P., William, N. C., Christel, A. S. B. and Christopher, J. H. P. : 50 years oral lipid-based formulations:
Provenance, progress and future perspectives. Advanced Drug Delivery Review. 101, 167(2016).
22) Abu, T. M. S., Salt formation to improve drug solubility.
Advanced Drug Delivery Reviews. 59, 603(2007).
23) Michael, P., John, D. H., Elizabeth, K., and Allen, C. T. : Strategies at the Interface of Drug Discovery and Development: Early Optimization of the Solid State Phase and Preclinical Toxicology Formulation for Potential Drug Candidates. Journal of Medicinal Chemistry, 53, 5897(2010).
24) Jin-Ju, B., Min-Ho, P., Seok-Ho, S. and Young-Geun, S. : Novel Lead Optimization Strategy Using Quantitative Structure-Activity Relationship and Physiologically-Based Pharmacokinetics Modeling. Yakhak Hoeji, 59, 151(2015).