논문 2011-3-11
포켓과 특징 점을 이용한 3차원 단백질 분자 형상인식
Shape Recognition of 3-D Protein Molecules Using Feature and Pocket Points
이항찬
* Hangchan Lee요 약
단백질 분자는 포켓 위치에서 유사한 형상을 갖는 다른 분자와 결합되며, 포켓은 단백질 분자의 형상을 묘사 하기 위한 참조 점으로 사용될 수 있다. Harris 검출기는 2 차원이나 3차원 객체의 특징 점을 검출하기 위해 널리 사 용된다. 특징 점들은 데이터의 변화율이 높은 영역과 포켓 영역에서 발견된다. 일반적으로 포켓 영역은 함몰된 형태로 존재하기 때문에 이 영역에는 다른 영역에 비해 다수의 특징 점들이 존재한다. 특징 점들을 포함하는 voxel cube를 연 속적으로 분할함으로써 포켓 영역을 발견할 수 있었고, 포켓 영역의 중심 좌표와 특징 점들 간의 Euclidean 거리를 계 산한 후 이들을 크기순으로 정렬 하였다. 정렬된 거리에 대한 그래프는 단백질 분자의 형상과 특징 점들의 분포에 대 한 정보를 제공하므로 단백질 분자를 형상별로 분리 할 수 있었다. 본 연구에서는 인위적인 잡음을 단백질 분자에 추 가하여 형상이 왜곡된 분자를 얻었고, 왜곡된 분자에 대해서도 95 % 이상의 정확 도로 형상을 인식 할 수 있었다. 정 확한 단백질 분자의 형상 인식은 분자들 간의 결합특성을 예측할 수 있는 중요한 정보를 제공한다.Abstract Protein molecules are combined with another ones which have similar shapes at pocket positions. The pocket positions can be good references to describe the shapes of protein molecules. Harris corner detector is commonly used to detect feature points of 2 or 3D objects. Feature points can be found on the pocket areas and the points which have high derivatives. Generally speaking, the densities of feature points are relatively high at pocket areas because the shapes of pockets are concave. The pocket areas can be decided by the subdivision of voxel cubes which include feature points. The Euclidean distances between feature points and the central coordinate of the decided pocket area are calculated and sorted. The graph of sorted distances describes the shape of a protein molecule and the distribution of feature points. Therefore, it can be used to classify protein molecules by their shapes. Even though the shapes of protein molecules have been distorted with noises, they can be recognized with the accuracy more than 95 %. The accurate shape recognition provides the information to predict the binding properties of protein molecules.
Key Words : Protein, Recognition, Harris Detector, Pocket.
I. 서 론
3차원 객체 인식은 원하는 객체를 데이터베이스로 부
*정회원, 한성대학교 멀티미디어 공학과 접수일자 2011. 4. 5, 수정일자:2011. 5. 30 개재확정일자 2011 6. 10
터 찾아내고 추출하는 일련의 과정을 말한다. 3차원 데이 터는 2차원에 비하여 데이터 량은 증가하지만 다량의 정 보를 정확하게 제공할 수 있다. 따라서 단순한 객체의 추 출뿐 아니라 단백질과 같은 생체 분자의 외형을 인식하 여 생화학적 특성을 예측하는 Bio-informatics의 한 분야 에도 적용할 수 있다[1]. 단백질 분자들은 서로 결합하여
특정한 구조와 특성을 가진 생물학적 개체로 분화하는데 이들의 결합은 유사한 외형을 가진 분자들끼리 결합할 확률이 높은 것으로 알려져 왔다[2]. 이에 따라 생물학적 실험을 통해 단백질 분자들의 결합특성을 연구해 왔으나 이를 위해서는 많은 시간과 용이 전제 되어야 한다. 최근 들어 컴퓨터를 이용한 3차원 객체 인식 알고리즘들이 개 발 되고 이는 일반적인 객체의 인식뿐 아니라 생명 공학 에도 응용되어 생물학적 실험에 소요 되는 막대한 비용 을 절감할 수 있는 토대를 마련하였다.
3차원 객체를 인식하는 알고리즘은 외형 분포에 의한 분류[3], 객체의 Entropy 에 의한 분류[4], vector 양자화에 의한 분류[5], voxel cube 에 의한 분류[6], 시각적 유사성에 의한 객체의 분류[7], 객체 표면을 구성하는 vector 분석에 의한 분류[8], 3차원 히스토그램에 의한 분류[9], 그리고 Fourier Transform 등 주파수 영역에서의 분류[10] 등 수 많은 알고리즘이 존재한다.
본 논문에서는 2차원 및 3차원 객체의 특징 점을 검출 하는데 널리 사용되는 Harris 검출기를 이용하여 단백질 분자의 외형을 분석하고 특징 점을 검출하였다. 검출된 특징 점들은 단백질 분자의 외형을 묘사할 수 있는 참조 점으로 사용될 수 있다. Voxelize된 단백질 분자에 Harris 검출기를 적용하였을 때 데이터의 변화율이 높은 영역의 중심 좌표가 특징 점으로 결정 되었고, 이러한 특 징 점들은 단백질 분자 간 상호 결합 위치로 알려진[11] 포 켓(pocket) 영역에 높은 밀도로 분포됨을 알 수 있었다.
포켓의 위치 및 형상은 단백질 분자 전체의 형상과 더불 어 단백질 분자의 결합 특성을 예측하기 위한 중요한 정 보를 제공한다. 본 논문에서는 포켓의 형상 보다는 단백 질 분자의 전체 형상 인식에 주안점을 둔다. 특징 점들을 검출한 후 이들의 밀도를 측정하여 포켓의 위치를 계산 하였고, 포켓의 중앙으로부터 나머지 특징 점들 사이의 Euclidean 거리를 측정하였다. 측정된 거리를 크기 별로 정렬하고 이를 토대로 단백질 간 형상의 유사도를 측정 하였다. 시스템의 강인성을 측정하기 위해 4개의 단백질 (1a7c_A, 1b8a_A, 1bef_A, 13pk_A)에 대해 voxel 좌표 값의 5~10% 에 해당되는 랜덤 잡음을 첨가한 후 원본 단 백질과의 유사도를 측정하였을 때 95.5% 의 높은 비율로 동종 단백질 분자의 형상을 인식할 수 있었다.
본 논문의 2장과 3장에는 각각 Harris 검출기와 포켓 검출 방법이 기술 되었고, 실험 결과 및 고찰은 5장에 주 어졌다.
Ⅱ. Harris Corner Detector
특징 점은 2차원 혹은 3차원 객체를 인식 하기 위한 기준점으로 널리 사용된다. Harris 검출기는 지역 데이터 분포를 사용하기 때문에 기하학적 변형에 대한 강인성을 가지고 있다[12][13]. Harris 검출기는 주어진 지점의 지역 autocorrelation 을 사용하여 특징 점을 결정 한다. 2차원 의 이미지를 고려할 때 주어진 한 점의 화소 값을
라 정의 하고 (Δx, Δy)에 의해 이동 되었다면 그 때의 autocorrelation 함수는 다음과 같이 정의 될 수 있 다. ∈
(1)
식(1) 에서 W(u, v) 는 한 점 (x ,y)를 중심으로 하는 윈도우로 정의 하고 두 번째 항인 는 Taylor 확장에 의해서 식 (2)와 같이 근사 될 수 있다.
≈
(2)
에 대한 편미분은
와
로 표현된다. 식 (1)의
항을 식(2)로 치환 하면 식(3)을 얻을 수 있다. ≈
∈
∈
(3)
주어진 영상의 데이터 분포 및 구조는 식(3)의 행렬
의 eigen-value
에 의해 설명 될 수 있 다. 즉 eigen-value
의 값이 크다면 특징 점이 될 수 있는 반면 작은 점은 평탄한 영역으로서 특징 점이 될 수 없다.지금까지 기술한 2차원의 Harris 검출기는 3차원 객체
의 특징 점 검출기로 사용될 수 있다. 식(3)에서의 2차원 행렬 T는 식(4) 와 같이 3차원행렬로 확장 될 수 있다 [12].
∈
(4)
행렬
에 대한 3개의 eigen-value를 구한 후 식(5) 와 (6)을 이용하여 determinant 와 trace를 계 산 하였다. 외부 변형에 대해 강인성을 보이는 sec 또한 식 (7)을 이용하여 얻을 수 있다. 마지막으로 주어진 지점 에 대한 특징 점의 강도(saliency)가 식(8)에 의해서 구해 진다. (5)
(6)
(7)
(8)
그림 [1~4]에서 각 그림의 좌측은 단백질 분자를 string으로 표현한 것이고 우측은 voxelize 된 단백질 분 자에 Harris 검출기를 적용하여 검출된 특징 점 중 강도 가 높은 50개의 특징 점들을 나타낸다.
50 100
40 150 60 80 100 120 140
160 1a7c-A
그림 1. String 으로 표현된 단백질 분자와 검출된 특징 점 들(1A7c_A)
Fig. 1. A protein molecule expressed by strings and feature points(1A7c_A)
50 100
40 150 60 80 100 120 140 160
1b8a A
그림 2. String 으로 표현된 단백질 분자와 검출된 특징 점 들(1b8_A)
Fig. 2. A protein molecule expressed by strings and feature points(1b8_A)
50 100
40 150 60 80 100 120 140
160 1bef-A
그림 3. String 으로 표현된 단백질 분자와 검출된 특징 점들(1bef_A)
Fig 3. A protein molecule expressed by strings and feature points(1bef_A)
50 100
150 40
60 80 100 120 140 160
13pk-A
그림 4. String 으로 표현된 단백질 분자와 검출된 특징 점들(13pk_A)
Fig 4. A protein molecule expressed by strings and feature points(13pk_A)
III. Pocket 검출 및 특징 점까지의 거리 산출
단백질은 크게 세 부위로 나뉜다. 아미노 말단 지역과 중앙 포켓지역, 그리고 카르복실 말단 지역이다. 이중 포 켓은 세포내의 여러 중요한 기능을 하는 단백질과 결합 하는 부위로서 단백질의 핵심 부위라고 할 수 있다. 이 포켓의 위치에서 어떤 단백질이 결합 하는가에 따라 다
양한 생물학적 특성을 갖는 생명체로 분화한다. 따라서 포켓의 위치와 단백질의 형상에 따라 어떠한 단백질 분 자가 결합 될 것인지에 대한 예측을 할 수 있고 이는 분 자 생물학에 있어서 중요한 관심 분야가 되고 있다. 본 논문에서는 단백질 분자의 형상을 인식하기 위한 기준점 으로써 포켓의 위치를 이용하고, 이에 대한 생물학적 언 급은 생략하기로 한다. 포켓의 형상은 단백질 마다 다르 나 다른 단백질과 결합을 위해 함몰된 형태로 존재하고 따라서 이곳에는 많은 특징 점들이 존재한다.
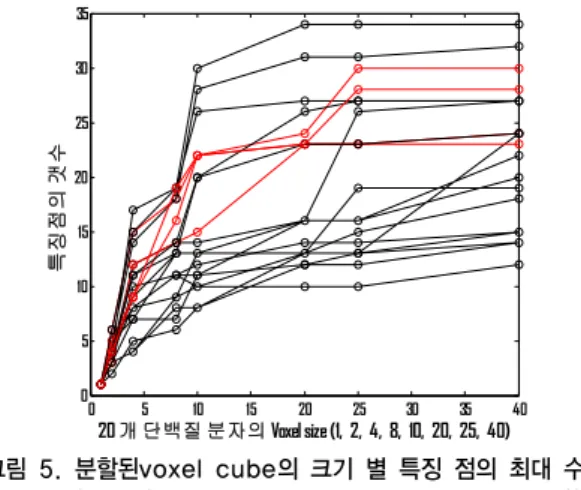
3차원의 voxel 형태로 주어진 단백질 분자의 데이터 분석을 위해, 200x200x200 데이터 중 특징 점이 존재하 는 위치에는 ‘1’을 삽입 하고 그렇지 않는 곳에는 ‘0’을 삽 입하였다. 각 단백질의 전체 voxel 데이터를 40x40x40, 25x25x25, 20x20x20, 10x10x10, 8x8x8, 4x4x4, 2x2x2 의 cube 로 분할하였을 때, 각 영역에 대하여 존재하는 특징 점의 개수를 측정하였고, cube의 크기 별 가장 밀도가 높 은 cube 대한 특징 점의 밀도를 그림5 에 표현 하였다.
실험 결과 분할된 voxel cube의 크기가 4x4x4일 때 포켓 의 중앙 위치를 가장 정확히 표현함을 알 수 있었다. 따 라서 4x4x4로 분할되었을 때 가장 밀도가 높은 voxel cube를 선택하고, 이 영역에 포함된 특징 점들의 좌표들 을 산술 평균하여 포켓의 위치로 결정하였다. 그림 [1~
4] 에서 볼 수 있는 것처럼 포켓의 위치에서는 많은 특징 점들이 밀집되어 있는 것을 알 수 있다. 포켓의 위치가 결정 된 후 포켓으로부터 모든 특징 점까지의 거리를 식 (9)에 의해 계산하였다. 여기서 n은 특징 점의 개수를 나 타내고
와
는 각각 포켓과 특징 점들의 좌표를 나타낸다.
(9)
식(9)에 의해 계산된 거리는 크기순으로 정렬되어 다 른 단백질 분자와의 유사성(correlation)을 측정하였고, 유사도에 따라 동종과 이종 단백질을 분리 할 수 있었다.
실험 결과는 4장에 주어진다. Harris 검출기를 이용하여 특징 점들을 검출하고, 이 특징 점을 포함 하는 200x200x200 voxel cube를 분할하여 포켓을 구한 후, 단 백질 인식까지의 전 과정은 그림6 의 블록 다이아 그램에 주어졌다.
0 5 10 15 20 25 30 35 40
0 5 10 15 20 25 30 35
20 개 단백질 분자의 Voxel size (1, 2, 4, 8, 10, 20, 25, 40)
특징점의갯수
그림 5. 분할된voxel cube의 크기 별 특징 점의 최대 수 (red: (1a7c_A, 1b8a_A, 1bef_A, 13pk_A)) Fig. 5. Maximum number of feature points for each size of voxel cube
Query Protein Molecule
Feature Point Detection
Pocket Detection
Distance Calculation
between Pocket and
Feature Points
Distance Sorting
Correlation Calculation and Recognition Harris
Detector Protein
Voxel Subdivision
Protein DB
그림 6. 단백질 형상 인식 시스템의 블록도
Fig. 6. Block diagram of the shape recognition system of protein molecules
IV. 실험 결과 및 토론
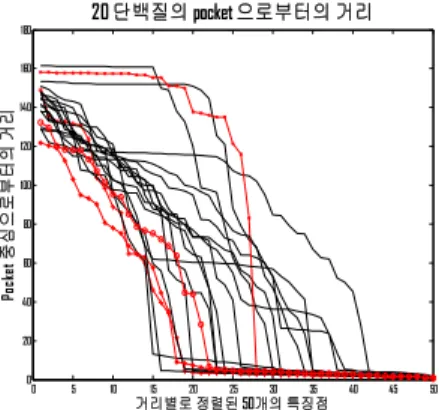
20개의 서로 다른 단백질 (1a7c_A, 1a88_A, 1aox_A, 1ar1_A, 1atk, 1auo_A, 1auy_A, 1aw1_B, 1aw5, 1ax4_A, 1ay0_A, 1aye, 1b8a_A, 1b77_A, 1b99_A, 1bar_A, 1bef_A, 1dzr_A, 1gwz, 13pk_A) 각각에 대해 포켓으로 부터 특징 점까지의 거리를 계산하고, 크기 별로 정렬한 후 그림 7 과 같이 그래프로 나타내었다. 그림7 에서 빨 간색과 (*, +, o, x)로 표현된 그래프는 시스템의 정확도 를 측정하기 위해, 잡음(noise)을 첨가 할 4개의 단백질 (1a7c_A, 1b8a_A, 1bef_A, 13pk_A)에 대한 거리를 나타 내고 있다. 즉 4개의 단백질에 대해서는 임의의 잡음을 첨가 한 후 형상 인식을 하였다. 그림7 에서 볼 수 있는 것처럼 각 단백질은 저마다의 형상에 따라 다른 형태의 그래프로 나타낼 수 있고 이들 그래프의 유사도를 측정 함으로써 단백질 분자의 형상을 인식할 수 있었다. 시스 템의 정확도를 측정하기 위해 4개의 단백질 분자의 voxel 좌표를 5 ~ 10%의 범위 내에서 난 수 발생기를
이용하여 임으로 이동하였다. 이는 자연 상태에서도 단 백질 분자는 내부적 혹은 외부적인 요인에 의하여 형상 의 일부가 변형될 수 있으므로 본 실험에서도 좌표 값을 변화시킴으로써 인위적으로 형상을 왜곡 시켰다. 즉 4개 의 단백질 분자에 대하여 범위 내의 잡음이 첨가된 50 개 의 왜곡된 단백질 분자를 생성한 후 그림6의 알고리즘을 적용하여, 포켓 과 특징 점간의 거리를 측정하였고 이를 그림8 에 표시하였다. 그림에서 빨간색과 (+) 기호는 왜 곡된 50개의 단백질 분자에 대한 포켓으로부터의 거리를 나타내고 있다. 그림에서 볼 수 있는 것처럼 분자의 형상 이 왜곡된 후에도 포켓으로부터 특징 점까지의 거리는 크게 변하지 않고 데이터가 유사한 위치에 모여 있음을 알 수 있었다. 이는 지역 영역에 기반을 투고 특징 점을 계산해 내는 Harris detector의 강점이기도 하다. 그림8 에서 특징 점 번호 20 이 후에 데이터가 모여 있는 것은 포켓 내부에 많은 특징 점들이 모여 있음을 시사한다.
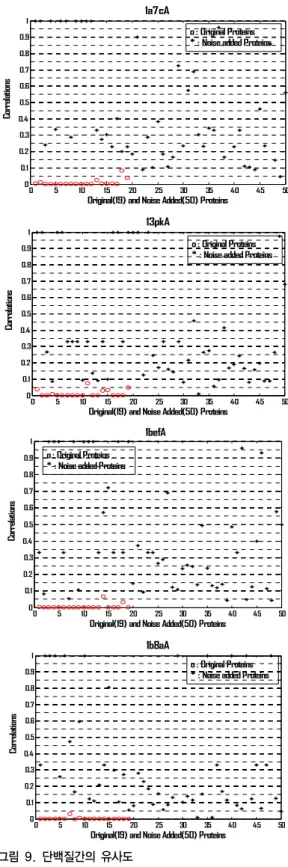
그림 9 는 잡음이 첨가된 단백질과 원본 단백질간의 correlation을 나타내고 있다. correlation이 0.05 이하인 경우는 인식에 실패한 것으로 간주한다. 그림에서 (*)로 표시된 것은 원본 단백질과 왜곡된 단백질 간의 correlation을 나타내고 (o)로 표시된 데이터는 실험에 사 용된 원본 단백질과 다른 19개의 이종 단백질간의 correlation을 나타낸다. 실험 결과 1a7c_A, 1bef_A, 13pk_A, 1b8_A의 경우 각각 98 %, 92%, 96%, 96% 의 인 식률을 보였다. 따라서 본 연구에서 사용한 4개의 단백질 분자에 대해서 평균 95.5 %의 높은 정확도로 형상을 인 식 할 수 있었다.
0 5 10 15 20 25 30 35 40 45 50
0 20 40 60 80 100 120 140 160 180
50
거리별로 정렬된 개의 특징점
Pocket 중심으로부터의거리
20 단백질의pocket 으로부터의 거리
그림 7. 20 개의 단백질 분자에 대한 포켓 중심과 특징 점들 간의 거리
Fig. 7. Distances between pocket and feature points for 20 protein molecules
0 5 10 15 20 25 30 35 40 45 50
0 20 40 60 80 100 120 140 160 180
50
거리별로 정렬된 개의 특징점
Pocket 중심으로부터의거리
Noise 첨가된 단백질의pocket 으로부터의 거리(1a7cA)
0 5 10 15 20 25 30 35 40 45 50
0 20 40 60 80 100 120 140 160 180
50
거리별로 정렬된 개의 특징점
Pocket 중심으로부터의거리
Noise 첨가된 단백질의pocket 으로부터의 거리(13pkA)
0 5 10 15 20 25 30 35 40 45 50
0 20 40 60 80 100 120 140 160 180
50
거리별로 정렬된 개의 특징점
Pocket 중심으로부터의거리
Noise 첨가된 단백질의pocket 으로부터의 거리(1befA)
0 5 10 15 20 25 30 35 40 45 50
0 20 40 60 80 100 120 140 160 180
50
거리별로 정렬된 개의 특징점
Pocket 중심으로부터의거리
Noise 첨가된 단백질의pocket 으로부터의 거리(1b8aA)
그림 8. Noise 첨가된 단백질의 포켓과 특징점 간의거리.
(red(+), 위로부터 1a7c_A, 13pk_A, 1bef_A, 1b8a_A)
Fig 8. Distances between pocket and feature
points for noise added protein molecules
0 5 10 15 20 25 30 35 40 45 50 0
0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
1 1a7cA
Original(19) and Noise Added(50) Proteins
Correlations
o : Original Proteins
* : Noise added Proteins
0 5 10 15 20 25 30 35 40 45 50
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
13pkA
Original(19) and Noise Added(50) Proteins
Correlations
o : Original Proteins
* : Noise added Proteins
0 5 10 15 20 25 30 35 40 45 50
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
1 1befA
Original(19) and Noise Added(50) Proteins
Correlations
o : Original Proteins
* : Noise added Proteins
0 5 10 15 20 25 30 35 40 45 50
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
1 1b8aA
Original(19) and Noise Added(50) Proteins
Correlations
o : Original Proteins
* : Noise added Proteins
그림 9. 단백질간의 유사도
(위로부터 1a7c_A, 13pk_A, 1bef_A, 1b8a_A) Fig. 9. Similarities among protein molecules
V. 결론
본 논문에서는 단백질 분자의 특징 점과 포켓에 기반 한 형상인식 알고리즘을 제안하였다. 즉 Harris 검출기를 이용하여 단백질 분자의 특징 점을 계산하고 특징 점들 의 단위 체적 당 밀도를 측정하여 포켓의 위치를 결정하 였다. 포켓으로부터 특징 점까지의 Euclidean 거리를 계 산하였고 이들을 크기 별로 정렬한 후, 비교 대상 단백질 데이터와 유사도(correlation)를 측정하였다. 제안된 알고 리즘의 강인성을 측정하기 위해 난수 발생기를 이용하여 단백질 분자의 voxel 좌표를 5 ~ 10 %의 범위 내에서 임의로 이동 시켜 잡음을 첨가 하였다. Harris 검출기는 지역영역 데이터 분포에 기반 하여 특징 점을 검출 하므 로 단백질 데이터에 약간의 잡음 혹은 변형이 추가 된다 고 하더라도 커다란 오차 없이 특징 점 들을 구해낼 수 있었다. 따라서 좌표 값의 이동에 의해 왜곡된 단백질 분 자에 대해서도 95.5 % 의 높은 인식률로 동종 단백질을 인식할 수 있었다. 생물학적인 측면에서 외형이 유사한 단백질끼리 서로 결합하는 특성을 가지고 있으므로 본 연구는 단백질 분자의 결합 특성을 예측하는데 유용하게 사용될 수 있을 것이고 이로 인하여 생물학적 실험에 소 요되는 비용을 절감할 수 있을 것으로 판단된다.
참고 문헌
[1] Rita Casadio, Gene Myers," Algorithms in Bioinformatics: 5th International Workshop WABI Mallorca, Spain, Oct. 3-6, Springer, 2005.
[2] James C. Whisstock and Arthur M. Lesk, Prediction of protein function from protein Sequence and structure, Quarterly Reviews of Biophysics vol. 36, pp. 307–340 March, 2003.
[3] Robert Osada et al, "Shape Distributions", ACM Transactions on Graphics, Vol. 21, No. 4, Pages
807–832. October, 2002.
[4] Benjamin Bustos, "Using Entropy Impurity for Improved 3D Object Similarity Search", IEEE International Conference onMultimediaandExpo (ICME),2004.
[5] Ryutarou Ohbuchi, “Salient Local Visual
※ 본 연구는 한성대학교 교내 연구비 지원 과제임.
Features for Shape-Based 3D Model Retrieval", Proc. IEEE International Conference on Shape Modeling and Applications (SMI’'08), Stony Brook University, June 4 - 6, 2008.
[6] Evgeny Ivanko and Denis Perevalov," Q- Gram Statistics Descriptor in 3D Shape Classification", LNCS 3687, pp. 360–.367, 2005.
[7] Ding-Yun Chen, Xiao-Pei Tian, Yu-Te Shen and Ming Ouhyoung, “On Visual Similarity Based 3D Model Retrieval“, EUROGRAPHICS Vol. 22, No.3, 2003.
[8] Ceyhun Burak Akgul,"Multivariate Density- Based 3D Shape Descriptors",IEEE International Conference on Shape Modeling and Applications SMI, 2007.
[9] Mihael Ankerst et al, "3D Shape Histograms for Similarity Search and Classification in spatial Databases ", Proc. 6th International Symposium on Spatial Databases (SSD‘'99), Hong Kong, China, July 1999. Lecture Notes in Computer Science.
[10] D. V. Vranic and D. Saupe, "3D Shape Descriptor Based on 3D Fourier Transform", CVSSP, pp. 271- 274. September, 2001.
[11] Brice Hoffman et al, “A new Protein binding pocket similarity measure based on comparison of clouds of atoms in 3D: application to ligand prediction”, BMC Bioinformatics, Nov., 2010.
[12] Fredrik Viksten, Klas Nordberg, and Mikael Kalms, “Point-of-Interest Detection for Range Data”, IEEE international conference on Pattern Recognition(ICPR), Dec., 2008.
[13] C. Schmid, R. Mohr, and C. Bauckhage, Evaluation of interest point detectors.
International Journal of Computer Vision, 37(2):151–172, June 2000.
저자 소개
이 항 찬(정회원)
∙1997년 미 오클라호마 주립대학교 졸 업(공박).
∙1997 ~ 1998 청운대학교 교수, 1999
~ 현재 한성대학교 교수
<주관심분야: 영상처리, 컴퓨터 비젼>