101
Stress-Induced Atherosclerosis: Clinical Evidence and Possible Underlying Mechanism
Ick-Mo Chung, M.D.
Division of Cardiology, Ewha Women’s University School of Medicine, Seoul, Korea
ABSTRACT
There is increasing recognition in medical fields of the importance of behavioral and psychosocial factors in the development of car- diovascular disease. Although the pathogenesis underlying stress-induced atherosclerosis is not well known, inflammation may play a key role. Activation of stress-induced neuroendocrine pathways, such as the hypothalamo-pituitary-adrenal axis, and the sympathe- tic nervous and renin angiotensin systems, direct neurogenic inflammation may also contribute to the development of stress-induced atherosclerosis. (Korean Circulation J 2005;35:101-105)
KEY WORDS:Stress;Atherosclerosis;Inflammation.
Introduction
Extensive studies support that behavioral and psychological factors contribute significantly to the development and preven- tion of atherosclerosis.1-4) Psychological factors, specifically depression, anxiety, personality factors, social isolation, and chronic and subacute life stress, are known to be related to the risk of coronary artery disease(CAD).2)
Psychological Stress is Estimated as an Emerging Risk Factor for Atherosclerosis
Stress can be defined as a threat to homeostasis provoked by a variety of stressor, such as environmental, psychological or physiological factors.1) Chronic stress has been linked to the development of insulin resistance and deposition of ab- dominal fat, risk factors for CAD and diabetes in both hu- mans and other primates.5) Social and psychological stress can provoke CAD in cynomolgus monkeys fed a low fat and low cholesterol diet, suggesting the possible role of stress in the development of atherosclerosis in people without traditional risk factors.4) Acute cardiovascular events and paradoxical arterial vasoconstriction are frequently triggered by physical
or mental stress in susceptible patients.6)7) In a large prospec- tive study, normotensive individuals, with high trait anger were at increased risk of combined CAD(acute myocardial infarction(MI)/fatal CAD, silent MI, or cardiac catheteriza- tion procedures) and hard events(acute MI/fatal CAD) compared with their lower anger counterparts.8) Similarly, the Framingham Heart Study demonstrated that suppressed anger independently predicted the 8-year incidence of CAD.9) Type A behavior pattern, a syndrome characterized by competition, hostility and exaggerated commitment to work, has received attention, because this personality trait is known to be associa- ted with a 2-fold increased risk of CAD and 5-fold increased risk of recurrent MI over an 8.5 year follow up.10) Conversely, other studies have reported no correlation between type A behavior and CAD.11) Therefore, some other confounding factors, such as socioeconomic status, seem to be important variables for the risk of CAD. Interestingly, hostility, a major attribute of the type A pattern, has been estimated as a potential toxic element in this personality type. The results of studies to assess the relationship between hostility and CAD have been mixed, suggesting certain components of the hos- tility construct are more pathogenic.2) Work-related stress is the most widely studied chronic life stress related to CAD.
Job strain, defined as jobs with high demand but low decision latitude, is associated with a 4-fold increase in the risk of cardiovascular death during a 6 year follow up,12) and when chronic, is positively associated with an increased risk of
Correspondence:Ick-Mo Chung, M.D.,Division of Cardiology, Ewha Wo- men’s University School of Medicine, 70 Jongro-6ga, Jongro-gu, Seoul 110-783, Korea
Tel:82-2-760-5076, Fax:82-2-762-7756 E-mail:ickmo@ewha.ac.kr
cardiovascular disease. Mental stress-induced myocardial is- chemia is known to be associated with a 2.8 fold higher rate of subsequent(mean 44 months follow up) fatal and nonfa- tal cardiac events, such as nonfatal MI, and coronary revascu- larization procedures.3) The role of stress in the progression of CAD has also been supported by the result of an interven- tion trial. Intensive lifestyle changes, including stress manage- ment, have been shown to significantly reduce angina attacks and adverse cardiac events, and reverse the degree of stenosis compared to a control group.13)
Physiological Pathways in Responding to Stress
Allostasis, defined as the ability to achieve stability through change, is critical to survival.5) Stress, as the allostatic load, can induce an allostatic response. The most common allosta- tic response involves the sympathetic nervous system(SNS) and hypothalamic-pituitary-adrenal(HPA) axis.5)17) Chemical mediators stimulated by stress activate the cells of paraventri- cular nucleus of hypothalamus(PVH) to produce corticotro- phin-releasing hormone(CRH), the key coordinator of stress.
CRH activates the corticotrophs of the anterior pituitary gland to produce adrenocorticotropic hormone(ACTH). CRH also stimulates the locus coeruleus to secrete norepinephrine(NE) at sympathetic nerve endings. Central activation of the SNS is transmitted to the adrenal medulla, where chromaffin cells are stimulated to produce epinephrine.
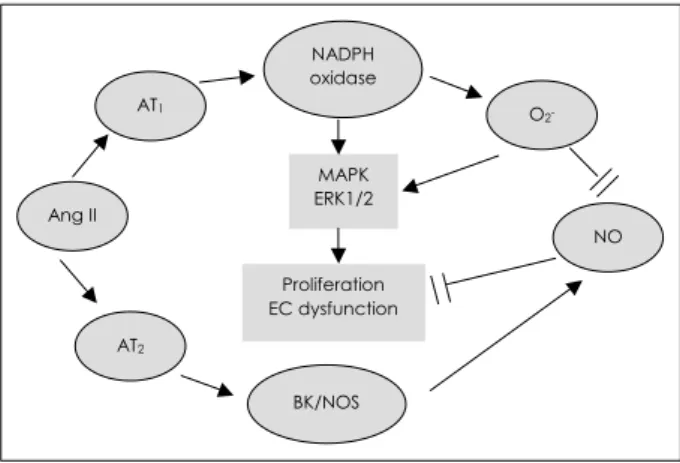
Stress can also activate the renin-angiotensin system(RAS), and central angiotensin receptors are important targets of stress regulation. Stress increases the production of circulating Ang II14) and the expression of its receptors in the brain, such as PVH.15) Stress-induced Ang II can activate the HPA axis through stimulation of hypothalamic CRH,16) and may con- tribute to the regulation of sympathetic responses to stress.15) Ang II has direct effects on the adrenal gland by stimulating aldosterone secretion from the zona glomerulosa and cate- cholamine release from the medulla.17) The effects of Ang II are mediated by two plasma membrane receptors, i.e. AT1
and AT2 subtypes.18) The majority of biological effects of Ang II occur following its binding to and activation of the AT1
receptor. There is crosstalk between AT1 and AT2 receptors, with stimulation of AT2 receptors opposing the effect of AT1
receptors(Fig. 1).19) Stress was found to produce significant increases in the AT1 receptor numbers and mRNA in PVH, AT1B receptor mRNA and AT1 receptor binding in the zona
glomerulosa of the adrenal gland and AT2 receptor mRNA in the locus coeruleus.15)20)
The effects of AT1 receptor antagonists on the stress-indu- ced hormonal and sympatho-adrenal system elucidate the role of RAS in stress pathophysiology. Blockade of AT1 receptors by an intracerebroventricular injection of Losartan inhibited the stress-induced increases in plasma catecholamine and CRH mRNA in the PVH.21) Peripheral administration of Candesar- tan, an AT1 receptor antagonist, which can cross the blood brain barrier, reverses the stress-induced increases in AT1B
mRNA in the adrenal gland zona glomerulosa, pituitary ACTH, and the secretions of adrenal corticosterone, aldoste- rone and catecholamine.15) These data suggest that endogenous Ang II induced by stress has a significant role in the activation of both the SNS and HPA axis.
Possible Mechanisms of Stress-Induced Atherosclerosis
Although much clinical evidence supports the concept of stress significantly contributing to CAD, the underlying me- chanism is not well known. Enhanced activation of the SNS can induce inflammation of the vasculature, leading to athe- rosclerosis, and can increase platelet adhesion and aggregation, hemostasis, thrombosis, lipid mobilization and activation of macrophages(Fig. 2).1)2) NE can control the release of CRH and enhance inflammation by stimulating the descending sym- pathetic signals. However, little is known of the biological pathways of neurogenic vascular inflammation. In addition to SNS activity, hypercortisolemia, resulting from the activation
NADPH oxidase
BK/NOS Ang II
AT2
AT1
NO O2-
MAPK ERK1/2
Proliferation EC dysfunction
Fig. 1. The role of angiotensin II (Ang II) mediated by receptor type I (AT1) and type II (AT2) in the arterial pathobiology. Note the opposite effects of AT1 and AT2. receptors. BK: bradykinin, NOS: nitric oxide synthase, NADPH: nicotinamide adenine dinu- cleotide phosphate, MAPK: mitogen activated protein kinase, ERK 1/2: extracellular signal-regulated kinase 1/2, NO: nitric oxide, EC: endothelial cell.
of the HPA axis, is associated with CAD.22)
Stress can induce inflammation in the peripheral organs via the neuro-endocrine system. It can be inferred that inflam- mation may act as a key mediator of stress-induced atherosc- lerosis. There are several inflammatory mediators contained within nerves that can be released by stress. These mediators include prostaglandin E2, neuropeptide Y(NPY), CRH, substance P(SP) and IL-6.1) NPY is a cotransmitter of sympathetic nervous innervation, which potentiates the action of NE, and can promote vascular smooth muscle cell prolife- ration, enhance leukocyte adhesion, platelet aggregation and macrophage activation.23) CRH may have a local direct effect on immune or inflammatory processes. Direct local adminis- tration of anti-CRH into the air pouch, simultaneously with carrageenin, an inducer of chemical inflammation, suppressed the inflammatory response to carrageenin, suggesting that CRH participates in the inflammatory process as a local stimulatory agent.24) Both somatic and autonomic nerves are associated with inflammatory cells, and nervous transmission induced by stress may result in neurogenic inflammation.1) Stress also increases the level of plasma proinflammatory
cytokines,25) and may induce macrophage, having β-adre- nergic receptors, to produce cytokines.26) One possible route to the stress-induced inflammation process is through the production of reactive oxygen species(ROS) subsequent to
Homocysteine Inflammation
Endothelial damage:
adhesion of platelets Stress Repeated acute or chronic
Susceptible, at risk person
Proinflammatory cytokines
Blood
Elevated blood pressure:
changes in blood flow
Liver:
acute phase proteins
Modification of lipid and cholesterol metabolism
Adhesion molecules
Progression of
inflammation Stable or
unstable plaques Plaque formation and progression:
activation of coagulation/fibronolysis
Stress hormones:
corticosteroids catecholamines
Recruitment of:
lymphocytes monocyres Corticosteroids
Shedding
Chemotactic molecules
Intimal mast cell activation
Fig. 2. Proposed mechanisms of stress-induced atherosclerosis.1)
Concentration of Ach (Log M)
Ach-7 Ach-6.5 Ach-6 Ach-5.5
Relaxation (%)
0 20 40 60 80
100 SD stress SD control
*
*
*
*
Fig. 3. Comparison of endothelial dependent vasorelaxation res- ponding to acetylcholine between rats subjected to 2 week im- mobilization stress and control group. SD: sprague-dawley, Ach:
acetylcholine. *: p< 0.05.27)
the activation of RAS. We found that arteries from rats sub- jected to stress showed endothelial dysfunction, as measured by the vascular tension in response to acetylcholine(Fig. 3).27) In our data, arteries of rats subjected to stress showed enhan- ced expression of endothelial adhesion molecules, such as vascular cell adhesion molecule(VCAM)-1 and intercellular adhesion molecule(ICAM)-1, decreased expression of en- dothelial nitric oxide synthase(e-NOS) mRNA and inacti- vation of nitric oxide(NO˙), compared with the control group.
Treatment of these rats with an ACE inhibitor significantly reversed both the stress-induced endothelial dysfunction and depression of e-NOS(unpublished data). Our data supports the hypothesis that “stress may inactivate NO˙, which is pro- bably mediated by the activation of RAS; thereby inducing vascular inflammation and endothelial dysfunction.
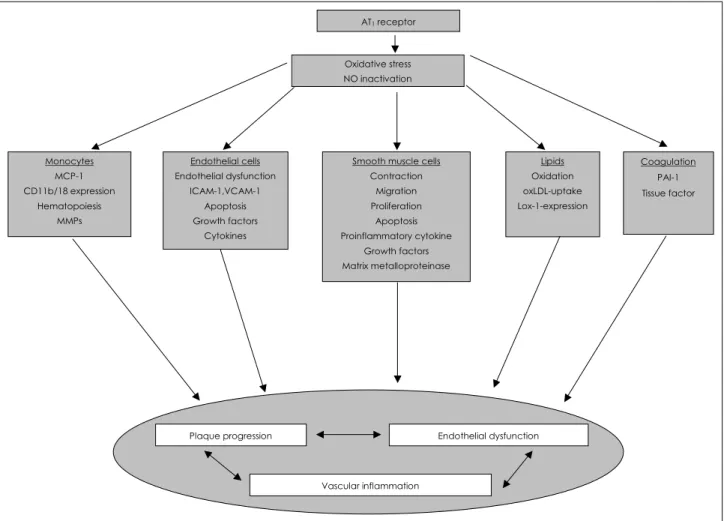
Ang II, via activation of AT1, has been implicated in cell migration, cell proliferation, coagulation, lipid oxidation and inflammation; thus, contributing to atherogenesis and endo-
thelial dysfunction(Fig. 4).18)28) AT1 receptor activation can lead to the production and release of ROS, such as superoxide (O2˙) and hydrogen peroxide(H2O2), in vascular cells, since AT1 receptors are linked to the activation of NADH/NADPH oxidase in vascular cells.29) However, other pathways, such as xanthine oxidase, uncoupling of e-NOS, lipoxygenase/cy- clooxygenase and Cyt P450 reductase, are known as sources of ROS production.19) When the production of ROS exceeds the ability for antioxidant defense, the resulting oxidant stress can evoke many pathophysiological conditions, such as dia- betes and atherosclerosis. Oxidant stress can activate redox sensitive genes, such as VCAM, ICAM and monocyte che- moattractant protein-1(MCP-1), thereby inducing inflamma- tion and progression of atherosclerosis.18) Increased superoxide production in response to Ang II can inactivate NO˙ by forming peroxynitrite(ONOO˙), which subsequently leads to endothelial dysfunction and promotion of atherosclerosis.
Peroxynitrite is a powerful oxidant and cytotoxic agent that
AT1 receptor
Oxidative stress NO inactivation
Plaque progression Endothelial dysfunction
Vascular inflammation Monocytes
MCP-1 CD11b/18 expression
Hematopoiesis MMPs
Endothelial cells Endothelial dysfunction
ICAM-1,VCAM-1 Apoptosis Growth factors
Cytokines
Smooth muscle cells Contraction
Migration Proliferation
Apoptosis Proinflammatory cytokine
Growth factors Matrix metalloproteinase
Lipids Oxidation oxLDL-uptake Lox-1-expression
Coagulation PAI-1 Tissue factor
Fig. 4. Contribution of AT1 receptor induced oxidative stress to atherosclerosis. AT1: angiotensin II type I, NO: nitric oxide, oxLDL: oxidized low density lipoprotein, MMPs: matrix metallo proteinases, PAI-1: plasminogen activator inhibitor, ICAM-1: intercellular adhesion molecule-1, VCAM-1: vascular cell adhesion molecule, MCP-1: mocyte chemoattractant protein-1.
can damage DNA, membrane lipids and mitochondria. Pero- xynitrite promotes the inflammatory synthesis of prostaglan- din30) and is responsible for activating cyclooxygenase. The product of cyclooxygenase, PGH2, is usually converted to PGI2 by prostacyclin synthase. However, prostacyclin syn- thase is susceptible to attack by peroxynitrite; therefore, the accumulation of PGH2 from cyclooxygenase can activate thromboxane receptors, resulting in vasoconstriction. This pathway may explain why susceptible patient complains of chest pain during stressful condition.
In conclusion, stress can induce atherosclerosis, which is probably mediated by inflammation. Various neuroendocrine pathways, such as the RAS and SNS, implicated in stress may participate in vascular inflammation. Future research to understand the mechanism of stress-induced atherosclerosis may elucidate the old curious association between mind and body.
REFERENCES
1) Black PH, Garbutt LD. Stress, inflammation and cardiovascular disease. J Psychosom Res 2002;52:1-23.
2) Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and impli- cations for therapy. Circulation 1999;99:2192-217.
3) Jiang W, Babyak M, Krantz DS, et al. Mental stress: induced myo- cardial ischemia and cardiac events. JAMA 1996;275:1651-6.
4) Kaplan JR, Manuck SB, Clarkson TB, Lusso FM, Taub DM, Miller EW. Social stress and atherosclerosis in normocholestero- lemic monkeys. Science 1983;220:733-5.
5) McEwen BS. Protective and damaging effects of stress mediators.
N Engl J Med 1998;338:171-9.
6) Muller JE, Tofler GH, Stone PH. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation 1989;79:
733-43.
7) Yeung AC, Vekshtein VI, Krantz DS, et al. The effect of atheros- clerosis on the vasomotor response of coronary arteries to mental stress. N Engl J Med 1991;325:1551-6.
8) Williams JE, Patron CC, Siegler IC, Eigenbrodt ML, Nieto FJ, Taylor HA. Anger proneness predicts coronary heart disease risk:
prospective analysis from the atherosclerosis risk in communities (ARIC) Study. Circulation 2000;101:2034-9.
9) Haynes SG, Feinleib M, Kannel WB. The relationship of psy- chological factors to coronary heart disease in the Framingham study: III. 8-year incidence of coronary heart disease. Am J Epi- demiol 1980;111:37-58.
10) Rosenman RH, Brand RJ, Jenkins D, Friedman M, Strauss R, Wurm M. Coronary heart disease in the Western Collaborative Group Study: final follow up experience of 8 1/2 years. JAMA 1975;233:872-7.
11) Regland DR, Brand RJ. Type A behavior and mortality from co- ronary heart disease. N Engl J Med 1988;318:65-9.
12) Karasek RA, Baker D, Marxer F, Ahlbom A, Thorell T. Job decision latitude, job demands, and cardiovascular disease: a prospective study of Swedish men. Am J Pbulic Health 1981;71:
694-705.
13) Ornish D, Scherwitz LW, Billings JH, et al. Intensive lifestyle changes for reversal of coronary heart disease. JAMA 1998;280:
2001-7.
14) Yang G, Xi ZX, Wan Y, Wang H, Bi G. Changes in circulating and tissue angiotensin II during acute and chronic stress. Biol Signals 1993;2:166-72.
15) Armando I, Carranza A, Nishimura Y, et al. Peripheral adminis- tration of an angiotensin II AT1 receptor antagonist decreases the hypothalamic-pituitary-adrenal response to isolation stress.
Endocrinology 2001;142:3880-9.
16) Aguilera G, Young WS, Kiss A, Bathia A. Direct regulation of hypothalamic corticotrophin releasing hormone neurons by an- giotensin II. Neuroendocrinology 1995;61:437-44.
17) Keller-Wood M, Kimura B, Shinsako J, Phillips MI. Interaction between CRF and angiotensin II in control of ACTH and adrenal steroids. Am J Physiol 1986;250:R396-402.
18) Nickenig G, Harrison DG. The AT1-type angiotensin receptor in oxidative stress and atherogenesis: I. oxidative stress and athero- genesis. Circulation 2002;105:393-6.
19) de Gasparo M. Angiotensin II and nitric oxide interaction. Heart Fail Rev 2002;7:347-58.
20) Dumont EC, Rafrafi S, Laforest S, Drolet G. Involvement of cen- tral angiotensin receptors in stress adaptation. Neuroscience 1999;
93:877-84.
21) Jezova D, Ochedalski T, Kiss A, Aguilera G. Brain angiotensin II modulates sympathoadrenal and hypothalamic pituitary adrenocor- tical activation during stress. J Neuroendocrinol 1998;10:67-72.
22) Kaplan JR, Petterson K, Manuck SB, Olsson G. Role of sympa- thoadrenal medullary activation in the initiation and progression of atherosclerosis. Circulation 1991;84(Suppl VI):VI23-32.
23) Zukowska-Grojec Z. Neuropeptide Y. a novel sympathetic stress hormone and more. Ann NY Acad Sci 1995;771:219-33.
24) Karalis K, Sano H, Redwine J, Listwak S, Wilder RL, Chrousos GP. Autocrine or paracrine inflammatory actions of corticotropin- releasing hormone in vivo. Science 1991;254:421-3.
25) LeMay LG, Vander AJ, Kluger MJ. The effects of psychological stress on plasma interleukin-6 activity in rats. Physiol Behav 1990;
47:957-61.
26) Coe CL, Rosenberg LT, Levin S. Prolonged effect of psychologi- cal disturbance on macrophage chemiluminescence in the squirrel monkey. Brain Behav Immun 1988;2:151-60.
27) Chung IM, Yoo MH, Han PL, Suh SH. Immobilization stress in- duced inflammatory change and dysfunction of arterial endothe- lium. Circ Res 2004;94:1523. Available from: URL: http://circres.
ahajournals.org/cgi/data/94/12/1523/DC1/1; Data supplement, page 8, abstr No. 24.
28) Libby P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation 2001;104:365-72.
29) Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griend- ling KK. Angiotensin II stimulation of NAD(P)H oxidase activity:
upstream mediators. Circ Res 2002;91:406-13.
30) Beckman JS. -OONO: rebounding from nitric oxide. Circ Res 2001;89:295-7.