A CASE OF OVARIAN CANCER DEVELOPED IN MAYER-ROKITANSKY-KÜSTER-HAUSER SYNDROME: INNATE CARCINOGENESIS OF OVARIAN CANCER
5
0
0
전체 글
(2) KJOG Vol. 55, No. 12, 2012. Fig. 1. A computed tomography scan shows rim-enhancing ovarian mass with solid part in pelvic cavity in patient with MayerRokitansky-Küster-Hauser syndrome.. 암으로 사망하였으며 이모가 유방암인 가족력이 있었다. 이학적 소견: 내진상 10년 전에 시술받은 질은 촉지되었으나, 질 입구 는 자궁과 연결되지 않았고, 흔적자궁은 만져지지 않았다. 우하복부에 약간의 압통과 반발통이 있었고, 거위 알 크기의 종괴가 만져졌다. 초음파 소견: 우측 부속기 쪽으로 9.4 × 6.7 cm 크기의 낭성 종괴(내부 에코와 유두모양, 고형부분을 지님)와 좌측 부속기 쪽으로 4.4 × 3.7 cm 크기의 혼합성 에코가 관찰되었다. 자궁은 3 × 4 cm 크기의 흔적자궁으 로만 발견되었다. 복부 및 골반 CT 소견: 우측 골반에 4 × 2 cm 크기의 유두 모양 부분을 포함한 8.5 cm 크기의 큰 낭성 종괴가 있고, 좌측 골반에 3 cm 크기의 고형 종괴와 3 cm 크기의 낭성 종괴가 있었다. 맹낭에 5 cm 크기의 고 형 종괴가 있었고, 이는 직장가(rectal shelf)로 추정되었다. 맹장 아래로 5 cm 크기의 종괴가 있었고, 간 주위로 저감쇄의 종괴들이 있었다. 비 정상적으로 커진 임파절은 없고, 폐 전이 소견은 없었다. 우측 난소의 낭성 선암으로 추정되었다(Fig. 1). 양측 신장과 요로는 정상 소견이었 다. 복부 및 골반 CT에서 관찰되는 척추의 기형은 없었다.. 1002. 수술 소견: 자궁 및 양측 부속기절제술, 양측 골반 임파술절제술, 부분 대망절제술, 잔류암 조직제거술을 시행하였다. 개복술 시 양측 흔적자 궁의 각 자궁각에서 정상적인 나팔관을 볼 수 있었다. 우측 난소는 남 성 주먹만한 크기의 종괴가 있었고, 종괴 안은 적갈색의 액체가 차 있 었고, 우측 골반벽과 유착되어 있었다. 좌측 난소는 거위알 크기의 종 괴가 있었고, 역시 적갈색의 액체가 차 있었고 S자 결장과 좌측 골반벽 과 유착되어 있었다. 상행 결장과 S자 결장, 직장에 거위알 크기의 종 괴가 있었고, 이를 제거하였다. 대망에는 전이된 소견은 없었으나, 부분 절제술을 시행하였고, 800-1,000 mL가량의 복수가 차 있었다. 병리 소견: 우측 나팔관과 붙어있는 9 × 6 × 3 cm 크기의 난소가 있었 고, 낭성 부분은 장액성 액체와 혈액으로 차 있었다. 고형 부분은 회색 과 황갈색의 부스러지기 쉬운 조직으로 되어있었다. 자궁은 3 × 1 × 1 cm 크기로 흔적자궁으로 남아있었고, 좌측 부속기는 4 × 3 × 1 cm 크기 였고, 출혈 낭종으로 보였다(Fig. 2). 흔적자궁이 수술에서 발견되어 환 자는 MRKH type II임이 확인되었다. 우측 난소에서 분화도가 낮은(grade III) 장액성 낭성 선암이 보이고. WWW.KJOG.ORG.
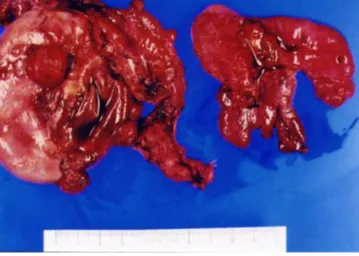
(3) A Ra Ko, et al. Innate carcinogenesis of ovarian cancer. Fig. 2. Right ovary contains solid nodule composed of gray tan friable tissue and multiple cysts which are filled with serous and hemorrhagic fluid. Two triangular small rudimentary uteruses are seen in the center.. 보이고, 좌측 골반내 4 cm 크기의 종괴 소견이 보였다. 재발성 난소암 으로 추정하여 종양감축술을 시행하였다. 우측 대장부분절제술, 비장 절제술, 부분대망절제술을 시행하였고, 비장, 우측 대장에 전이된 종괴 및 간 표면과 장간막, 대장에서 다발성 소결절들이 확인되었다. 조직 소견에서 실질부위 비장에 1.4 × 0.8 cm 크기, 비문부위의 5.5 × 4 cm 크기, 우측 부분절제술(우측 상행 결장, 회장, 맹장, 충수 부위의 장막과 비장막) 부위의 4.5 × 3.5 cm 크기의 악성 종괴가 확인되었다. 또한 떼 어낸 모든 조직(맹장, 충수, 우측 대장, 회장 말단, 비장, 장간막)에서 전 이된 장액성 낭성 선암 소견이 관찰되었다. 재수술 후 이차 항암요법 (carboplatin, area under curve (=4) × (glomerular filtration rate + 25); taxol, 135 mg/m2; gemcitabine, 800 mg/m2)을 시행하였으나 6차례 치료 후에도 복막 암종증이 지속되어 환자는 더 이상의 치료를 거부하 였고 진단 및 수술 후 48개월째에 난소암 진행에 의한 다발성 장기부 전으로 사망하였다.. 고찰. Fig. 3. Serous carcinoma of the ovary illustrates poor differentiation (H&E, ×100).. (Fig. 3), 흔적자궁 주위로 전이된 암세포가 확인되었다. 망과 S자 결장 및 대장 주위에서 떼어낸 조직에서도 암세포가 확인되었다. 골반 임파 선의 전이는 없었다. 면역조직화학검사(immunohistochemical stain)상 p53은 명확치 않았고, Ki-67은 양성 소견을 보였다. 최종병기는 3기말 (IIIc)로 결정되었다. 수술 후 경과: 수술 후 환자의 경과는 비교적 양호하였으며, 수술 후 복합항암화학요법(cisplatin, 75 mg/m2; taxol, 135 mg/m2; 24시간 점 적)을 6차례 시행하였다. 항암요법 후 암표지자 추적관찰 결과 cancer antigen (CA) 125는 600 U/mL 이상에서 6.4 U/mL로, CA 15-3는 69 U/mL에서 16 U/mL으로 감소하였고, CT 결과 또한 전이나 재발 소견 은 없었다. 1차 항암요법 10개월 후 암표지자인 CA 125는 6.4 U/mL에서 323 U/mL로 증가하였고, CT상 간과 비장 주위로 복막의 다발성 소결절이. WWW.KJOG.ORG. 뮬러관 발육부전은 뮬러관의 마지막 부위의 발달 실패에 의한 것으 로 뚜렷한 질이 없고, 원발성 무월경이 있는 경우 진단된다[3]. 질형성 부전은 여성 출생 4,500명 중 한 명 꼴로 발생하는 드문 여성 생식기 발달 기형이다. 이러한 환자들은 질이 없거나 얕은 질 입구를 가진 질 의 부분적 결손을 보이고, 자궁은 흔히 없다. 하지만 드물게 자궁이 정 상인 경우가 있으나, 이런 경우 질과의 연결통로가 미약하거나 흔적자 궁 혹은 쌍 각의 코드로만 남아있다. 난소는 뮬러관에서 발생하는 장기 가 아니기 때문에 난소의 기능은 정상이고, 따라서 정상적인 이차 여성 성징을 갖고 성장을 하게 된다[1]. MRKHs의 경우 type I과 II로 분류되는데 다른 동반기형이 없고, 상 부 2/3의 질과 자궁이 전혀 형성되지 않았을 경우를 type I MRKHs 로 진단하고, 흔적자궁이 남아있거나, 상부요로기관, 척추, 손발가락, 귀 등의 다른 기관에 동반기형이 있는 경우에는 모두 type II MRKHs 라고 한다. 특히 동반기형이 있는 경우를 Müllerian duct aplasia, renal dysplasia and cervical somite anomalies association, genital renal ear syndrome라고 칭하기도 한다[4]. Type II MRKHs과 관련있는 기형들은 비뇨기계통의 기형, 골격계의 기형[5] 그리고 중이 기형과 난청[6]이다. 특히 비뇨기계 기형은 발병률 이 40%에 이르고, 단일 신, 마제신, 골반내 신장, 중복 신우와 요관 등 으로 나타난다[4]. 골격계의 기형은 10%-12%에서 발견되고, 척추골 기형이 흔하고, 과잉 혹은 비대칭적 척추 등도 나타나며, 또한 KlippelFeil 증후군과도 관련이 있다고 보고되고 있다[5]. 마지막으로 뮬러관 형성부전은 중이 기형과 난청과도 관련이 있으며, 그 발생률은 Strübbe 등[6]의 연구에 따르면 25%에 이르는 것으로 보고된다. 이 증후군의 원인은 아직 밝혀지지 않았고, 대부분의 경우 산발적으 로 발생하지만, 몇 가족에서 발생한 예를 보고한 바가 있다[7]. 이러한 소수의 가족에서 발생한 유전 양식으로는 상염색체 열성 유전의 패턴. 1003.
(4) KJOG Vol. 55, No. 12, 2012. 처럼 보인다[8]. 이 증후군과 관련있다고 보고된 원인 중 anti-Müllerian hormone (AMH), anti-Müllerian hormone receptor (AMHR) [9], 유전 자의 돌연변이로 인한 AMH의 과다 노출로 인한 원인일 수 있다는 가 설이 있었으나, 이후 이 염색체들이 원인적 역할을 하지 못할 것이라는 연구보고도 있다[10]. 또한 galactose-1-phosphate uridyl transferase 유전자의 N314D 다형성과의 관련성이 있을 것이라는 많은 연구가 있 었고, galactose 대사의 오류로 인한 galactose의 자궁내 노출의 증가 가[11] 확실한 원인으로 추정되었으나, 이 염색체의 다형성도 큰 관련 이 없다는 최근 연구보고가 있다[12]. 이에 따라 이 증후군의 원인에 대해서는 아직 확실히 알려진 바가 없고, 유전적 요인과 환경적 요인을 포함한 다인성 요인이 작용할 것으로 생각된다. 청소년기에 발생하는 원발성의 무월경이 이 증후군을 진단하게 하 는 임상적 주요 특징이다. 따라서 흔히 뮬러관 형성부전은 청소년기 에 일차성 무월경에 대한 조사과정에서 진단된다. 진찰상 자궁이 촉지 되는 경우 구조물의 대칭성과 크기에 대해 조사하기 위해 초음파를 실 시하지만 초음파상에서 확실하지 않은 경우 자기공명영상(magnetic resonance imaging, MRI)검사를 시행해야 한다[13]. MRI가 초음파보 다 정확하고 복강경에 비해 비용이 저렴하면서 덜 침습적이므로 주로 사용하고, 진단적 복강경은 흔히 필요하지 않지만, 흔적자궁의 자궁근 종, 혈자궁, 자궁내막증, 증상이 있는 탈출증과 같은 병변이 있을 시는 복강경수술을 고려한다. 또한 조사과정에는 신우촬영, 뼈와 귀, 코, 목 에 대한 검사가 기형 감별을 위해 필요하다[11,12]. 또한 원발성 무월 경을 주증상으로 하는 터너증후군과 안드로겐무감응증후군(androgen insensitivity syndrome)과의 감별이 필요한데, MRKH증후군은 정상적 인 핵형, 즉 46, XX를 갖는 반면에 터너증후군에서는 난포자극 호르몬 (follicle stimulating hormone)의 상승과 45, XO의 핵형이 특징적이며, 안드로겐무감응증후군에서는 46, XY의 핵형과 난소, 자궁 대신 고환이 존재하며, 음모와 액와모의 발달이 없거나 미미한 것이 차이점이다. 또 한 질결손과 구분해야 할 것으로 처녀막막힘증(imperforated hymen), 가로질중격(transverse vaginal septum) 등이 있다. 정상적인 성관계를 갖지 못하는 것과 관련하여 기능적인 새로운 질 을 만들어 주는 수술을 한다. 여러 기술요법이 알려져 있고, 그들 중 질 강내 부분층 피부 이식술을 이용한 McIndoe 수술법과 Frank의 비수술 적 요법, William의 질성형술, Creatsas 변형법, Vecchietti의 수술법 등 들이 현재 널리 시행되고 있다. 기능적인 질 생성수술의 성공으로 뮬러 관 형성부전 환자들에게 정상적인 성적 생활을 가능하게 하고 이 증후 군에 따른 고통을 완화시킬 수 있다. 그러나 의사들은 이러한 환자들이 정상적으로 발달된 난소와 나팔관 그리고 다양한 크기의 두 개의 흔적자궁이 있을 수 있다는 것과 뮬러 관 형성부전 환자들을 대할 때 골반내 병적 질환의 가능성과, 하복부에 서 발생하는 증상에 대해 부인과적 조사가 필요하다는 것을 명심해야 한다. 실제로 MRKHs 환자에서 다낭성 난소증후군이 발견된 바가 여러 차례 발표되었고[14] 드물게 미분화 기형종이나 내배엽동 종양 등 악 성 종양을 동반한 경우가 증례보고된 바 있다[15]. 본 예에서는 환자의 나이가 발병 당시 37세로 젊은 나이에 발생한. 1004. 난소암이며, 암 가족력을 가지고 있으므로, 유전적 원인이 발병에 주된 영향을 미쳤으리라 추론할 수 있다. MRKH 증후군 환자에서 생긴 난소 암은 질을 통한 외부와의 공간적 차단이 이루어진 상태에서 발생한 것 이므로 외부인자의 영향 없이 유전자 변이만으로도 난소암이 발생할 수 있다는 개념의 난소암 발암기전(ovarian cancer carcinogenesis)을 보여주는 좋은 모델이다. 이 증례 외에서도 MRKH 증후군 환자에서 난 소암이 병발한 경우가 드물게 보고되고 있어[2], 추가적 증례 수집을 통하여 MRKH 증후군 환자에서 난소암이 발생하는 내인적 요인을 밝 히고자 하는 관심이 필요하다.. References 1. Capraro VJ, Gallego MB. Vaginal agenesis. Am J Obstet Gynecol 1976;124:98-107. 2. Ghirardini G, Magnani A. Mayer-Rokitansky-Kuster-Hauser syndrome and ovarian cancer. Report of a case. Clin Exp Obstet Gynecol 1995;22:247-8. 3. Griffin JE, Edwards C, Madden JD, Harrod MJ, Wilson JD. Congenital absence of the vagina. The Mayer-Rokitansky-KusterHauser syndrome. Ann Intern Med 1976;85:224-36. 4. Morcel K, Camborieux L; Programme de Recherches sur les Aplasies Müllériennes, Guerrier D. Mayer-Rokitansky-KusterHauser (MRKH) syndrome. Orphanet J Rare Dis 2007;2:13. 5. Willemsen WN. Combination of the Mayer-Rokitansky-Kuster and Klippel-Feil syndrome: a case report and literature review. Eur J Obstet Gynecol Reprod Biol 1982;13:229-35. 6. Strübbe EH, Cremers CW, Dikkers FG, Willemsen WN. Hearing loss and the Mayer-Rokitansky-Kuster-Hauser syndrome. Am J Otol 1994;15:431-6. 7. Shokeir MH. Aplasia of the Mullerian system: evidence for probable sex-limited autosomal dominant inheritance. Birth Defects Orig Artic Ser 1978;14:147-65. 8. Wottgen M, Brucker S, Renner SP, Strissel PL, Strick R, Kellermann A, et al. Higher incidence of linked malformations in siblings of Mayer-Rokitansky-Kuster-Hauser-syndrome patients. Hum Reprod 2008;23:1226-31. 9. Resendes BL, Sohn SH, Stelling JR, Tineo R, Davis AJ, Gray MR, et al. Role for anti-Mullerian hormone in congenital absence of the uterus and vagina. Am J Med Genet 2001;98:129-36. 10. Zenteno JC, Carranza-Lira S, Kofman-Alfaro S. Molecular analysis of the anti-Mullerian hormone, the anti-Mullerian hormone receptor, and galactose-1-phosphate uridyl transferase genes in patients with the Mayer-Rokitansky-Kuster-Hauser syndrome. Arch Gynecol Obstet 2004;269:270-3.. WWW.KJOG.ORG.
(5) A Ra Ko, et al. Innate carcinogenesis of ovarian cancer. 11. Cramer DW, Goldstein DP, Fraer C, Reichardt JK. Vaginal agenesis (Mayer-Rokitansky-Kuster-Hauser syndrome) associated with the N314D mutation of galactose-1-phosphate uridyl transferase (GALT). Mol Hum Reprod 1996;2:145-8. 12. Klipstein S, Bhagavath B, Topipat C, Sasur L, Reindollar RH, Gray MR. The N314D polymorphism of the GALT gene is not associated with congenital absence of the uterus and vagina.. Mol Hum Reprod 2003;9:171-4. 13. Pompili G, Munari A, Franceschelli G, Flor N, Meroni R, Frontino G, et al. Magnetic resonance imaging in the preoperative assessment of Mayer-Rokitansky-Kuster-Hauser syndrome. Radiol Med 2009;114:811-26. 14. Appelman Z, Hazan Y, Hagay Z. High prevalence of mullerian anomalies diagnosed by ultrasound in women with polycystic. Mayer-Rokitansky-Küster-Hauser syndrome 환자에서 발병된 난소암 한림대학교 의과대학 산부인과학교실. 고아라, 김지현, 박수예, 전영은, 김성주, 임채춘, 강정배, 박영한 질무형성증은 여자 신생아 4,000명 중 1명에서 나타나는 드문 선천 생식기 기형이다. 대부분의 경우에서 Mayer-Rokitansky-KüsterHauser syndrome (MRKHs) 증후군에 속한다. 이 증후군을 가진 환자는 질이 없으며 자궁은 없거나 맹관을 형성하고 있으나, 난소는 정상적 으로 기능하여 2차 성징이 정상으로 나타난다. 본원에서 MRKH증후군 여성에서 발생한 난소암의 드문 증례를 경험하여 보고하는 바이다. 중심단어: MRKH 신드롬, 난소암, 질무형성증. WWW.KJOG.ORG. 1005.
(6)
수치
관련 문서
