ARTICLE Vol. 3, No. 2, November 2010
논문접수일: 2010년 4월 30일 / 심사완료일: 2010년 8월 10일
교신저자: 송민호, 대전시 중구 대사동 640번지, 301-721, 충남대학교병원 내과
Tel: 042-280-7161, Fax: 042-280-7995, E-mail: [email protected] 위 종설은 2009년 젠자임학술상 수상기념 논문입니다.
갑상선유두암의 병인에 근거한 치료제의 개발과 문제점
충남대학교 의학전문대학원 내과학교실
이민희, 조영석, 송민호
Development of New Therapeutics Based on Pathogenesis of Papillary Thyroid Cancer
Min Hee Lee, MS, Young Suk Jo, MD, PhD and Minho Shong, MD, PhD
Laboratory of Endocrine Cell Biology, Division of Endocrinology, Department of Internal Medicine, Chungnam National University School of Medicine, Daejeon, Korea
Papillary thyroid cancer (PTC) is the most common endocrine malignancy. The prognosis of PTC is defined primarily by the principal clinical features such as patient’s age, tumor size, histological subtypes, regional nodal metastasis, extra-thyroidal invasion and distant metastasis. Although PTC prognosis is excellent, adverse clinical features increase the probability of recurrence and progression. To improve patients’ survival and reduce recurrence and progression, advances in molecular biological insights for principal clinical features will beneeded. It has been recognized for years that aberrant kinase activation can induce cellular transformation, carcinogenesis and disease progression. Based on this concept, many investigators have been focusing on the development of new small molecules to inhibit the aberrant kinase activation and conducted clinical trials.
In this review article, we describe the background for development of new targeted therapy and define molecular biological insights into the carcinogenesis of PTC.
Key Words: Papillary thyroid cancer, BRAF
V600E, Sorafenib, PLX4720
서 론
갑상선암의 발병률은 지난 20년간 전 세계적으로 50% 이상 증가 되고 있으며 특히 여성에서 현저하게 증가하고 있다.
1)국내에서도 급격한 증가를 보이며, 2004년 이래 한국 여성에서 흔한 발생률을 보이는 암 으로 보고되고 있다(보건복지가족부 중앙암등록본부 2008년 10월 15일 발표 자료).
갑상선암의 치료법으로는 수술과 방사성 요오드, 갑 상선 자극호르몬(TSH) 억제 등이 사용되고 있으며, 이 러한 치료법에 의해서 분화갑상선암의 10년 생존율이 90% 이상이 되었다. 그러나 40년 동안 장기 추적조사 에서 약 35% 환자들이 재발하였으며, 해마다 미국지역
의 1,600명이, 전 세계적으로 35,000이 갑상선암에 의해 사망하고 있다.
미분화형 또는 요오드 흡수력이 없거나 탈분화가 된 갑상선암은 방사성 요오드 치료에 반응성이 낮으며, 따라서 높은 재발율과 불량한 예후를 나타낸다. 예후 가 나쁜 암에 대하여 현재 사용되는 치료법들은 효과 가 적고, 또한 치료반응이 오래 지속되지 못하는 단점 을 지니고 있다.
1)갑상선암은 여포상피세포로부터 유도되는 분화갑상 선암(DTC)이 95%로 가장 많으며, 수질암(MTC) 4%와 역형성형암이 1%로 뒤를 잇는다. 분화갑상선암 중 유 두암(PTC)이 80%, 여포암(FC)이 10%, 휘틀세포암이 5%를 차지한다. BRAF
V600E변이는 PTC에서 60∼80%
까지 발견되는 가장 흔한 유전적 변이로써 이 변이의
발견은 갑상선암을 진단할 수 있는 대표적인 분자 표 지자이다.
BRAF
V600E변이는 PTC를 유발하는 가장 중요한 유
전자 변이로서 암 발생과 진행 과정에 관여할 것으로 예상된다. 따라서 BRAF
V600E변이에 의한 비정성적으 로 증가된 키나아제 활성을 억제할 수 있는 소분자 화 합물 개발은 치료적 관점에서 매우 중요한 연구분야이 다 .
2)본 논문에서는 갑상선암의 발생에 관련된 oncogenic kinases의 작용을 억제하는 치료법의 개발에 대한 배경 을 이해하기 위하여 BRAF 키나아제의 특성, BRAF 억 제제의 현재까지의 연구와 치료제 개발의 한계점을 지 적하고자 한다.
RAF 동형과 야생형(wild type) BRAF의 활성
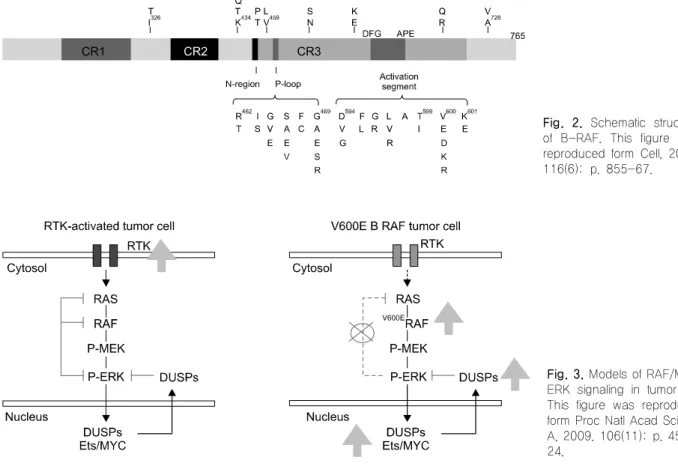
Serine-threonine RAF 키나아제는 포유류에서 ARAF, BRAF, CRAF/RAF-1 세 개의 형태로 존재하며, 공통적 으로 RAS 결합부위를 포함한 CR1, CR2와 키나아제 도 메인인 CR3로 구성된다(Fig. 1). ARAF가 약 68 KD이 고 CRAF가 약 74 KD이며, BRAF는 엑손 8b과 엑손 10a의 대체접합(alternative splicing) 등에 의해 크기가 67에서 99 kD로 다양하다. 또한 대체접합은 BRAF가 MEK를 활성화 시키는 능력에 영향을 주는데 엑손 10a 가 포함되었을 때 기저 키나아제 활성이 증가되는 반 면 , 엑손 8b가 포함되었을 때에는 기저 키나아제 활성 이 감소한다.
3)또한, RAF 동형들은 조직 발현차이가 생기는데 CRAF는 대부분의 조직과 세포에서 광범위
하게 발현되며, BRAF는 조혈세포, 신경 등에서 발현되 며, 갑상선 여포세포에서 일부 발현된다.
4)야생형 BRAF는 활성 루프와 ATP 결합 부위와의 소 수성 결합을 하고 있어 불활성 상태를 유지한다.
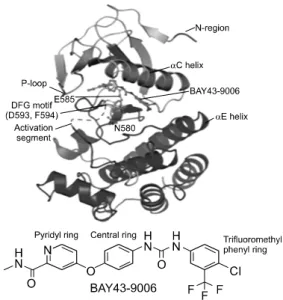
5)또 한 , 타이로신 키나아제 수용체(RTK)와 RAS 활성에 의 하여 CRAF (RAF-1)와의 복합체를 이루어 MEK, ERK 활성을 유도하는 기전을 지니고 있다(Fig. 1).
6)RAF 복합체는 mlk (mixed lineage kinases 3)-BRAF- CRAF (RAF-1) 복합체가 알려져 있다(Fig. 1). mlk는 BRAF의 인산화에는 영향을 주지 않지만, BRAF와 CRAF의 heterodimer 안정화에 기여한다. 현재 mlk- BRAF-CRF 복합체에 대한 정확한 구성은 알려져 있지 않으나 CNK1 (connector enhancer of KSR1), Sur-8 (suppressor of ras-8) 등과 같은 chaperone과 KSR1, EP1, Paxillin과 같은 scaffold proteine 등이 포함되어 있다.
7)암에서의 BRAFV600E 키나아제의 발현
BRAF 변이는 악성 흑색종, 대장암, 자궁암, 갑상선 암 등에서 보고되고 있다. 전체 갑상선암의 90% 이상 을 차지하는 분화 갑상선암에서 발현되는 BRAF변이 의 대부분은 N lobe의 glycine-rich P 루프와 활성분절 (activation segment) 부분에서 집중 발생된다.
갑상선암에서는 BRAF K601E, V600E, V600D, K601del, V599ins, FGLAT601-605ins 및 AKAP9 BRAF rearrangement와 같은 점 돌연변이, 삽입과 삭제 그리 고 유전자 재배열에 의해 BRAF의 oncogenic 활성화에 기여한다.
1)BRAFV600E는 BRAF 유전자의 15번 exon 의 1799번 뉴클레오타이드의 티미딘이 아데노신으로 치환되어 600번 아미노산인 발린이 글루탐산로 치환된 변이이다. 지금까지 보고된 30개 이상의 단일변이 중 90%를 차지하며, PTC에서 60∼80%까지 보고된 가장 일반적인 유전적 변이이다(Fig. 2).
BRAF
V600E변이는 BRAF의 활성분절 인산화 위치인
Thr599와 Ser602이가 인산화된 야생형 BRAF와 유사한 구조를 가지게 된다. 결과적으로 이러한 구조적인 특 성으로 BRAF
V600E변이는 야생형 BRAF와 달리 소수성 결합이 붕괴되고, 기저 키나아제 활성이 증가되며, Ras 의 관계없이 MEK-ERK 경로를 활성화 한다.
5,8)BRAF에 의해서 활성화된 ERK는 vascular endothelial
growth factor (VEGF) 같은 혈관생성 인자의 발현을 증
가시켜 종양 형성을 유도한다. 또한 ERK는 인산화를
통하여 CREB (cAMP responsive element binding pro-
Fig. 3. Models of RAF/MEK/
ERK signaling in tumor cell.
This figure was reproduced form Proc Natl Acad Sci U S A, 2009. 106(11): p. 4519- 24.
Fig. 2. Schematic structure of B-RAF. This figure was reproduced form Cell, 2004.
116(6): p. 855-67.
tein)를 활성화하고 세포사멸 유도인자인 BAD (BCL-2 antagonist of cell death), Bim (BCL-2-interacting mediator of cell death)을 억제하거나, 캐스페이즈를 비활성화시 켜 세포사를 회피한다.
9-11)ERK는 FOS, cyclin D1, DUSP4, DUSP6 등과 같은 전사인자들을 발현시키며, DUSP6, IGFBP7 등과 같은 하위의 인자의 피드백시스템에 의하여 조절 받는다.
그러나 BRAFV600E 변이는 ERK를 억제하는 다양한 피드백시스템을 저하시켜 ERK의 과활성을 유도한다 (Fig. 3).
12,13)결과적으로 BRAFV600E 돌연변이와 변이에 의한 ERK의 활성화가 종양의 발생과 성장에 중요한 역할을 하는 것으로 규명됨에 따라 BRAF를 표적으로 하는 다 양한 억제화합물들이 개발 되었다.
BRAFV600E의 억제 화합물
현재 다양한 화합물들이 RTK-RAS-RAF-MEK-ERK 기전과 RAS 하위인자에 관여하는 억제제로 약 43종 개 발 되었으며, 약 38종의 억제제의 임상시험이 진행되고
있다.
14,15)임상시험이 시행되고 있는 약제 중, 갑상선암
발현에 주요 인자인 BRAFV600E의 억제제로 개발되어
임상시험 중인 약제로 BAY 43-9006 (sorafenib), PLX4720/
PLX4032, RAF-265, XL281이 있다.
BAY 43-9006 (sorafenib)
BAY 43-9006 (sorafenib)의 작용 기전
암 유전자들을 표적으로 하는 키나아제 약물들이 많 이 개발되고 있으며, imatinib, sunitinib, vandetanib, sorafenib 등의 Tyrosine 키나아제 억제제들이 알려져 있다. 이들 중 BRAF 억제제인 BAY43-9006 (Sorafenib) 은 BRAF와 CRAF (RAF-1) 같은 RAF 키나아제들을 억 제할 뿐만 아니라 VEGFR 1-2-3, RET, RET/PTC, PDGFR 같은 tyrosine 키나아제도 억제한다(Fig. 4, Table 1).
17)또한 sorafenib은 경구투여가 가능하다는 장점이 있지 만, 변이 BRAF보다는 야생형 BRAF에서 억제 효과가 높다는 단점이 있다.
8,17)임상시험
다양한 RAF 키나아제에 작용하는 sorafenib은
BRAFV600E 돌연변이 암에 대한 치료 기대 효과가 있
기에 갑상선암과 흑색종에서 임상시험이 진행되었고
Fig. 4. Binding of B-RAF and BAY43-9006. This figure was reproduced form Cell, 2004. 116(6): p. 855-67.
Table 1. Biochemical IC50 of BAY43-9006
Assay IC50. nM
CRAF 6±3
BRAF 22±6
BRAFV600E 38±9
VEGFR-2 90±15
Fit-3 58±20
C-KIT 68±21
FGFR-1 580±100
ERK-1, MEK-1, >10,000
EGFR, IGFR-1
This table was reproduced form Cancer Res, 2004. 64(19):
p. 7099-109.
최근 그 결과들이 발표 되었다.
갑상선암에서는 Gupta-Abramson 등과 Kloos 등에 의해 임상시험이 시행되었으며, Gupta-Abramson 등은 전이성이고 요오드 불응성인 갑상선암 환자에게 sora- fenib 400 mg을 하루 2회 경구 투여하는 phase II를 진 행하였다 . 7명의 환자들에서 부분관해(PR)가 18주에서 84주까지 지속되었으며, 16명의 환자들에서는 14주에 서 89주까지 안정질환(SD)을 보였다.
18)Kloos 등도 sorafenib 400 mg을 하루 2회 경구 투여하는 phase II를 진행하였다. 41명의 PTC 환자들 중 6명은 PR이 관찰 되었으며, 23명의 환자들에서는 SD가 6개월 이상 지속 되었다 . 18명의 Tg (thyroglobulin)-평가 환자 중 14명은 25% 이상 Tg가 감소하였다.
19)흑색종에서는 McDermott 등과 Hauschild 등에 의해 임상시험이 진행되었는데 McDermott 등은 dacarbazine (DTIC) 50명과 sorafenib+dacarbazine 51명, 총 101명 의 환자를 대상으로 phase II를 시행하였다. PFS (pro- gression-free survival)는 DTIC 그룹은 11.7주 sorafenib
+dacarbazine 그룹은 21.1주였다(ASCO). Hauschild 등 은 unresectable III기 IV기 흑색종환자 270명을 대상으 로 항암제로 알려져 있는 Carboplatin와 Paclitaxel (CP) 를 Placebo 그룹과 sorafenib 그룹에 같이 투여하는 방 식으로 phase III를 시행하였다. Placebo+CP 135명과 sorafenib+CP 135명으로 나뉘어 Paclitaxel은 225 mg/m
2정맥주사로, sorafenib은 400 mg을 경구투여로 진행하였다. PFS는 Placebo+CP 그룹은 17.9주, sorafenib+CP 그룹은 17.4주였다. Placebo+CP 그룹의
15명의 환자에서 PR이, 51%인 69명의 환자에서 SD가 관찰되었으며, sorafenib+CP 그룹의 16명의 환자가 PR 을 54%인 79명이 SD를 보였다.
20,21)PLX4720/4032
PLX4720의 작용 기전
BRAFV600E 특이적인 억제제의 개발의 필요에 따라 Raf265 (Novartis), XL281 (Exelixis/Bristol Myers Squibb), AZ628 (AstraZeneca), SB-590885 (GlaxoSmithkline) and PLX-4720 (Plexxikon/Roche) 같은 소분자 억제제들이 개발 되었으며, RAF 키나아제들을 억제하는 sorafenib 보다는 좀 더 특이적으로 인식하는 약물들이다. 특히 PLX4720/PLX4032는 BRAF 특이적인 소분자 억제제로 BRAF V600E에 대한 IC
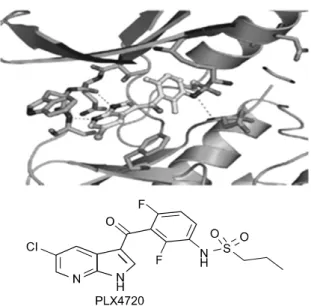
50값이 13 nM로 야생형 BRAF 와 10배 이상의 차이를 지니며 특이적으로 BRAF 변이 를 억제하는 효과를 지닌 약물이다 (Fig. 5, Table 2).
21)PLX4720은 hinge 부위에 가까이 있는 키나아제 domain의 N과 C lobe 사이에 갈라진 부위에 결합하면 서 ATP 결합 위치를 막는다.
22)PLX4720의 프로필 그룹은 RAF의 활성형태에 선택
적으로 결합하는데 중요하다. 활성화된 BRAFV600E
변이와 야생형 BRAF는 DFG 형태를 공유하지만, αC
helix의 위치에 차이가 발생하며, αC helix의 Glu-501
은 catalytic Lys-483로부터 구분되기 때문에, PLX4720
은 독특한 구조로 발암성의 BRAF 키나아제를 억제 한
다 . 대조적으로, large tail 그룹이 있는 BAY43-9006 같
은 화합물은 표적 물질이 오직 비활성 형태로 존재 할
때만 선택 가능한 포켓에 결합한다 . 이 선택 가능한 포
켓은 많은 키나아제들에 보존되어 있어서, “일반적인
Fig. 5. Structures of PLX4720. This figure was reproduced form Proc Natl Acad Sci U S A, 2008. 105(8): p. 3041-6.
Table 2. Biochemical IC50 of PLX4720
Assay IC50. nM
BRAF V600E 13
BRAF 160
BRK 130
FRK 1,300
CSK 1,500
KDR 2,300
AURORA A 3,400
This table was reproduced form Proc Natl Acad Sci U S A, 2008. 105(8): p. 3041-6.
키나아제 포켓”이라고 불린다. 즉, PLX4720는 야생형 BRAF와 BRAFV600E 변이 모두의 형태들과 결합 할 수 있지만, 활성형태를 선호하는 특성과 RAF-선택적인 포켓에 대한 결합은 non-RAF 키나아제와의 비교하여 중요한 특성으로 작용한다.
20)임상시험
PLX4032는 현재 전이성 흑색종에서 임상시험이 진 행 중이나, MEK 억제제들과 비교하여 주목할만한 성 과는 보고되지 않고 있다 . BRAF 변이 양성환자만 포함 된 phase I은 PLX4032 960 mg을 하루 2회 경구투여 하 였고, 31명 중 15명이 50%가 넘는 종양억제를 18명은 30%가 넘는 억제를 보였다. Plexxikon은 지난 9월 100 명의 환자를 대상으로 phase II 시작하였고, 700명의 환 자를 대상으로 흑색종에서 PLX4032와 dacarbazine (DTIC-Dome)를 비교하는 phase III를 시작하였다 (ASCO). 그 밖에 RAF265는 전이성 흑색종에서 1회 복 용량을 증가하는 방식으로 안전성, 약물 동태학, 약력 학을 평가하는 phase I/II를 진행하고 있으며, XL281은 폐암, 결장암, 갑상선유두암, 흑색종에서 phase I으로 1 일 1∼2회 경구투여로 안전성과 최대투여용량(MTD) 을 연구하고 있다(http://www.clinicaltrials.gov/).
BRAF V600E 억제화합물의 문제점
BRAF 억제화합물들은 임상시험 단계에서 치료제로 의 한계점들이 관찰되었다. BRAF를 특이적으로 인식
하지 못하고, tyrosine 키나아제 등의 비특이적 억제효 과를 보이는 BAY 43-9006은 세포성장에 영향을 줄 수 있기 때문에 치료제로서의 한계점이 있다. 또한 특정 인자를 인식하지 않은 비특이적 억제제이므로 심혈관 질환, 위장관질환 등과 갑상선 기능 저하증의 부작용 이 우려되고 있다.
16)갑상선암에서 행한 두 종류의 임 상시험에서 피로, 피부발진, 무기력, 고혈압 등의 부작 용이 나타났으며,
18,19)흑색종에서 행한 임상시험에서 도 피부발진과 설사, 피로 등의 부작용이 관찰되었
다.
20,21)BRAF
V600E를 특이적으로 인식하는 PLX4720은
전이성 흑색종 환자를 대상으로 한 phase I에서 피로, 발진, 골통 등이 관찰되었으며, 환자의 23%에서 편평 상피세포암 , 각질극 세포종이 발달하였다(ASCO).
최근 Heidorn 등,
23)Hatzivassiliou 등,
24)Poulikakos 등
25)은 RAF 억제제가 지니는 특이적인 문제점을 제시하였
다 . 위 세 개의 논문에서는 공통적으로 흑색종 세포를
가지고 실험을 진행하였으며 BRAF
V600E변이가 세포에
서는 억제제가 RAF-MEK-ERK를 강력하게 억제하는
반면에 RAS변이가 있는 세포에서는 MEK/ERK 신호를
증가시킨다고 주장했다. Heidorn 등은 KRAS 변이와
RAS/RAF 야생형 tumor에서 선택적인 BRAF inhibitor
인 885-A는 BRAF-CRAF 복합체 형성을 촉진하여서
CRAF에 의한 MAPK 활성화를 촉진한다고 주장하였
다 . 또한 Heidorn 등은 inhibitor에 의한 효과가 없더라
도 ‘kinase-dead’ BRAF는 CRAF와의 복합체가 유도되
는 관찰하였다고 보고하였다.
23,26)Hatzivassiliou 등은
GCD-0879에 의해 BRAF-CRAF, CRAF-CRAF의 복합
체 형성이 촉진되며, CRAF 의존적으로 억제제로서 작
용한다고 주장한다. PLX4720은 BRAF-CRAF의 복합체
형성은 억제하지만, CRAF-CRAF의 복합체 형성을 유
도한다고 발표했다 .
24,26)Poulikakos 등은 sorafenib, PLX4720,
JAB34 등의 억제제는 RAS 변이를 가지고 있는 종양세
포에서뿐만 아니라 , HER2 같은 종양형성 유전자에 의 해 활성화 되는 세포에서도 MEK와 ERK를 활성화 한 다고 밝혔다.
25,26)세 개의 논문들은 다른 RAF 억제제를 사용하였으며, 억제제가 RAF-MEK-ERK 기전의 활성화에 어떠한 영 향을 주었는지 규명하는 방법도 다르다 . 그러나 BRAFV600E 변이에 의한 암은 과거 밝혀진 연구 결과와 동일하게 BRAF V600E를 억제하지만 RAS 돌연변이에 의해 발 생되는 암은 억제하지 못하는 것을 동일하게 주장하고 있다 . 이 논문들은 BRAF 억제제가 역설적으로 암 세포 의 성장을 촉진할 수 있는 기전을 제시하였으며, 약제 시험 과정에서 나타난 부작용을 설명할 수 있는 계기 가 되었다.
요약 및 결론
갑상선암을 비롯한 여러 암에서 비정상적인 키나아 제들에 의하여 암이 발생하고 진행 및 재발에 관여하 는 것이 알려져 있다. 악성흑색종, 대장암, 자궁암, 갑 상선암에서 발견된 BRAF
V600E변이는 RTK/RAS에 의 한 활성과 관계없이 ERK의 활성을 일으키고 유지하는 기전이 밝혀졌다. 그래서 BRAF
V600E/MEK/ERK 기전을 억제하는 화합물들이 암 치료제로서의 효과를 보일 것 으로 기대되고 연구되었다 . 하지만, 다중키나아제 억제 제는 경구투여가 가능하다는 이점이 있으나, 특이적으 로 암 유전자를 인식하지 못하기에 세포 성장을 억제 하는 문제점을 확인하였다 . 암 유전자를 특이적으로 인식하는 억제제의 개발의 필요성이 제시됨에 다양한 소분자 화합물들이 개발되었다.
PLX4720/PLX4032같은 소분자 억제제들은 BRAF
V600E변이를 특이적으로 억제한다는 이점이 있으나 , RAS변 이와 RAS/RAF 야생형 종양에서 암 유발을 향상시킨다 는 부작용이 보고되면서 특이적인 억제제나 치료제로 서의 문제점이 제기되었다 . 치료제로서의 부작용이 제 기 됨에 따라 다른 억제제들과 혼용하여 사용하면서 단일 억제제의 사용에 따른 부작용을 해결하기 위한 노력이 계속되고 있다 . BRAF 억제치료가 나타내는 문 제점의 생물학적 측면의 기전을 규명하는 것도 효능이 높은 치료제개발을 가능케할 것으로 생각된다.
중심 단어: 갑상선유두암, BRAFV600E, Sorafenib, PLX4720.
References
1) Tang KT, Lee CH. BRAF mutation in papillary thyroid
carcinoma: Pathogenic role and clinical implications. J Chin Med Assoc 2010;73(3):113-28.
2) Schreck R, Rapp UR. Raf kinases: Oncogenesis and drug
discovery. Int J Cancer 2006;119(10):2261-71.
3) Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is
identified as a mutational target. Biochim Biophys Acta 2003;
1653(1):25-40.
4) Gotz R, Wiese S, Takayama S, Camarero GC, Rossoll W, Schweizer U, et al. Bag1 is essential for differentiation and
survival of hematopoietic and neuronal cells. Nat Neurosci 2005;8(9):1169-78.
5) Nikiforov YE. Thyroid carcinoma: molecular pathways and
therapeutic targets. Mod Pathol 2008;21(Suppl 2):S37-43.
6) MacCorkle RA, Tan TH. Mitogen-activated protein kinases in
cell-cycle control. Cell Biochem Biophys 2005;43(3):451-61.
7) McKay MM, Morrison DK. Integrating signals from RTKs to
ERK/MAPK. Oncogene 2007;26:3113-21.
8) Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK
signaling pathway by oncogenic mutations of B-RAF. Cell 2004;116(6):855-67.
9) Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK
signaling pathway by transcription-dependent and -independent mechanisms. Science 1999;286(5443):1358-62.
10) Jo YS, Li S, Song JH, Kwon KH, Lee JC, Rha SY, et al.
Influence of the BRAF V600E mutation on expression of vascular endothelial growth factor in papillary thyroid cancer. J Clin Endocrinol Metab 2006;91(9):3667-70.
11) Erhardt P, Schremser EJ, Cooper GM. B-Raf inhibits
programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol Cell Biol 1999;19(8):5308-15.
12) Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB,
et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A 2009;
106(11):4519-24.
13) Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR.
Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008;
132(3):363-74.
14) Fecher LA, Cummings SD, Keefe MJ, Alani RM. Toward a
molecular classification of melanoma. J Clin Oncol 2007;25(12):
1606-20.
15) Montagut C, Settleman J. Targeting the RAF-MEK-ERK
pathway in cancer therapy. Cancer Lett 2009;283(2):125-34.
16) Licitra L, Locati LD, Greco A, Granata R, Bossi P.
Multikinase inhibitors in thyroid cancer. Eur J Cancer 2010;
46(6):1012-8.
17) Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral
antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004;64(19):7099-109.
18) Gupta-Abramson V, Troxel AB, Nellore A, Puttaswamy K, Redlinger M, Ransone K, et al. Phase II trial of sorafenib in
advanced thyroid cancer. J Clin Oncol 2008;26(29):4714-9.
19) Kloos RT, Ringel MD, Knopp MV, Hall NC, King M, Stevens R, et al. Phase II trial of sorafenib in metastatic thyroid
cancer. J Clin Oncol 2009;27(10):1675-84.
20) Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al.
Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A 2008;
105(8):3041-6.
21) Hersey P, Bastholt L, Chiarion-Sileni V, Cinat G, Dummer R, Eggermont AM, et al. Small molecules and targeted therapies
in distant metastatic disease. Ann Oncol 2009;20(Suppl 6):vi35-40.
22) Hauschild A, Agarwala SS, Trefzer U, Hogg D, Robert C,
Hersey P, et al. Results of a phase III, randomized, placebo-
controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unrese- ctable stage III or stage IV melanoma. J Clin Oncol 2009;27(17):
2823-30.
23) Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu- Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic
RAS cooperate to drive tumor progression through CRAF. Cell 2010;140(2):209-21.
24) Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to
activate the MAPK pathway and enhance growth. Nature 2010;464(7287):431-5.
25) Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N.
RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464(7287):427-30.
26) Cichowski K, Janne PA. Drug discovery: Inhibitors that activate.