REVIEW ARTICLE
염증성 장질환과 Inflammasome
김정목
한양대학교 의과대학 미생물학교실
Inflammatory Bowel Diseases and Inflammasome
Jung Mogg Kim
Department of Microbiology, Hanyang University College of Medicine, Seoul, Korea
Inflammatory bowel disease (IBD), the most important entities being ulcerative colitis and Crohn's disease, are chronic, relapsing and remitting inflammatory conditions that result from chronic dysregulation of the mucosal immune system in the intestinal tract. Although the precise pathogenesis of IBD is still incompletely understood, increased levels of proinflammatory cytokines, including interleukin (IL)-1β, IL-18 and tumor necrosis factor-α, are detected in active IBD and correlate with the severity of inflammation, indicating that these cytokines may play a key role in the development of IBD. Recently, the intracellular nucleo- tide-binding oligomerization domain-like receptor (NLR) family members, including NLRP1, NLRP3, NLRC4 and NLRP6, are emerging as important regulators of intestinal homeostasis. Together, one of those aforementioned molecules or the DNA sensor absent in melanoma 2 (AIM2), apoptosis-associated speck-like protein containing ‘a caspase recruitment domain (CARD)’
(ASC) and caspase-1 form a large (>700 kDa) multi-protein complex called the inflammasome. Stimulation with specific microbial and endogenous molecules triggers inflammasome assembly and caspase-1 activation. Activated caspase-1 leads to the secretion of proinflammatory cytokines, including IL-1β and IL-18, and the promotion of pyroptosis, a form of phagocyte cell death induced by bacterial pathogens, in an inflamed tissue. Therefore, inflammasomes are assumed to mediate host defense against microbial pathogens and gut homeostasis, so that their dysregulation might contribute to IBD pathogenesis. This review focuses on recent advances of the role of NLRP3 inflammasome signaling in IBD pathogenesis. Improving knowledge of the inflammasome could provide insights into potential therapeutic targets for patients with IBD. (Korean J Gastroenterol 2011;58:300-310) Key Words: Inflammasomes; Inflammatory bowel diseases; Intestinal epithelial cells
CC This is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/
by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
교신저자: 김정목, 133-791, 서울시 성동구 행당동 17번지, 한양대학교 의과대학 미생물학교실
Correspondence to: Jung Mogg Kim, Department of Microbiology, Hanyang University College of Medicine, 17 Haengdang-dong, Sungdong-gu, Seoul 133-791, Korea.
Tel: +82-2-2290-0645, Fax: +82-2-2282-0645, E-mail: [email protected]
Financial support: This work was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Educa- tion, Science, and Technology (MEST) (2010-0008594) and a grant from the National Research Foundation of Korea funded by the Korean Government (MEST) (MRC Program 2010-0029507). Conflict of interest: None.
서 론
병원균이 인체에 감염되면 1차 방어기전으로 선천면역반 응(innate immune response)이 작동한다. 이 때 제일 처음 반응하는 것은 침입한 병원균의 pathogen-associated mo- lecular pattern (PAMP)을 숙주세포에 있는 pattern recog- nition receptors (PRRs)로 인지하는 것이다. PRRs을 통한 시 그널은 다양한 염증 경로, 즉 mitogen-activated protein kinase (MAPK), caspase-1, nuclear factor (NF)-κB, acti-
vator protein-1 (AP-1) 등을 활성화시켜 염증반응을 유도한 다.
PRRs의 대표적인 예로, 세포막 표면에 위치하는 toll-like receptor (TLR)와 세포질 내부에 존재하는 nucleotide-bind- ing oligomerization domain (Nod)-like receptor (NLR)를 들 수 있다.1-3 이 PRRs은 인체 조직 또는 세포가 파괴되면서 유출되는 damage-associated molecular patterns (DAMPs) 도 인지할 수 있다.4 즉, TLRs은 세포질막과 endosome의 바 깥 표면에 위치하는 반면, NLRs은 세포질 내부에 위치하면서
세포 내부로 들어온 PAMPs와 DAMPs을 인지한다. 따라서 이들 PRRs은 장내 미생물무리(enteric microbiota)와 장조직/
세포 유출물질을 감지하여 장내 항상성(gut homeostasis) 유 지에 기여한다.5,6 예를 들어 PRRs은 장관의 병원균 정착 (colonization)과 감염을 조절하고, 장벽기능의 유지, 손상된 상피세포의 회복, 그리고 장점막 내부의 면역반응의 균형 유 지 등을 수행할 수 있는 시그널을 촉발하는 역할을 한다.6-9 그러므로 PRRs 기능이 왜곡될 경우 장관 기능손상으로 질환 이 초래될 가능성이 대단히 높다.
염증성 장질환(inflammatory bowel disease, IBD)의 병인 은 아직까지 명확하게 밝혀지지 않고 있으나, 다양한 환경 요 인들이 장점막의 면역반응을 왜곡시킴으로써 초래되는데, 이 때 유전적 감수성이 있는 개체에서 질병 발현이 높아진다는 것이 일반적으로 정립된 개념이다.10,11 환경 요인의 대표적인 예로 장내 미생물무리의 조성 변화를 들 수 있다.12,13 즉, IBD 에서는 장내 미생물무리의 조성이 변화되어 있고 장점막에 부 착된 균수가 증가되어 있다.14,15 따라서 장내 미생물무리에 대 한 잘못된 면역반응이 IBD 발생에 기본을 이룰 것이라는 이 론이 제시되고 있다.12,13,16,17
장상피세포(intestinal epithelial cells, IECs)로 구성된 장 벽(IEC barrier)은 장관 내부에 존재하는 장내 미생물무리로 부터 점막조직을 보호한다. 건강한 개체에서는 비병원성 공생 균(commensal)이 해로운 면역반응 없이 장관 안에서 살게 허락하는, 즉 면역관용(immune tolerance)의 상태로 존재한 다. 그러나 유전적 감수성이 있는 개체에서는 장벽 기능이 제 대로 발휘되지 못하기 때문에, 공생균이 점막 내부로 침입하 게 된다. 침입한 미생물은 TLR과 NLR 같은 PRRs을 통하여 대식세포(macrophage), 수지상세포(dendritic cell), 호중구 (neutrophil) 등의 면역세포와 반응한다. 이와 같은 PRRs의 활성은 친염증성 매개체(proinflammatory mediator; cyto- kine, chemokine, prostaglandin 등)의 발현을 촉진함으로 써 면역반응을 가속화시키고, 그 결과 IBD 발생으로 이어진 다.10,18
2002년 Martinon 등19은 NLR의 subset 중 NLRP1 (또는 NALP1, CARD7)을 포함한 다수의 분자들이 결합되어 cas- pase-1 신호전달 경로를 활성화시키고, 그 결과 친염증성 cy- tokine인 interleukin-1β (IL-1β)와 IL-18이 분비된다는 결과 를 발표하였다. 이후 통상적으로 NLRP1이 포함된 multi-mo- lecular complex를 ‘NLRP1 inflammasome’이라고 부르고 있 다. 한편 NLRP3 (또는 NALP3, cryopyrin, CIAS1, PYPAF1, CLR1.1)와 NLRC4 (또는 IL-1β converting enzyme [ICE]- protease activating factor [IPAF])도 inflammasome의 구 성분자로 발견되었다. 최근에는 비-NLR type인 ‘absent in melanoma 2 (AIM2)’라고 명명된 pyrin and HIN (PYHIN)
domain을 포함하는 protein family가 포함된 inflam- masome (AIM2 inflammasome)과 NLRP6가 포함된 in- flammasome (NLRP6 inflammasome)도 보고되고 있다.20,21 이중에서 NLRP3가 장관의 항상성을 조절하는 대표적인 인자로 떠오르고 있다. 예를 들어, NLR family member인 NLRP3 유전자의 single nucleotide polymorphism (SNP) 연구를 통해 NLRP3가 크론병과 연관되어 있다는 보고는 NLRP3 inflammasome의 중요성을 보고한 사례라고 할 수 있다.22 앞서 소개한 바와 같이 NLRP3는 inflammasome을 구 성하는 세포 내 단백이다.23-25 따라서 NLRP3가 포함된 in- flammasome (NLRP3 inflammasome)은 IL-1β와 IL-18의 생성을 유도한다. 이 cytokine들은 염증을 유발할 뿐 아니라 상처가 난 IECs 회복에도 관여한다는 점에서,26 NLRP3 in- flammasome은 IBD의 질병 조절에 관여할 가능성이 대단히 높다. 그러나 이와는 반대로 NLRP3 inflammasome이 IBD 발 병 및 진행을 촉진한다는 보고들도 있다.27,28 한편 NLRP6 in- flammasome도 장관 내부의 정상균무리와 장염 조절에 관여 한다는 최근의 보고로 미루어,28 다른 형태의 inflamma- some도 IBD 병인론에 관여할 가능성이 높다. 이 종설에서는 현재까지 보고된 inflammasome 중에서 주로 NLRP3 in- flammasome과 IBD 병인론과의 관련성에 대하여 고찰해 보 고자 한다.
본 론
1. IBD에서의 NLR 시그널의 중요성
NLRP3와 관련된 NLR 단백인 Nod1과 Nod2는 장내 미생 물무리에 대한 염증반응을 조절하는 중요한 인자이다. Nod1 과 Nod2는 장점막에 있는 IECs, Paneth cell, 항원제시세포 (antigen-presenting cells, APCs)에서 핵전사인자(nuclear transcription factor)인 NF-κB와 AP-1을 활성화시킨다.17 그 런데 IBD 환자의 약 15-20%에서 Nod1과 Nod2를 coding하 고 있는 유전자의 돌연변이가 관찰되고 있다.29-32 또한 Nod1 과 Nod2는 autophagy (또는 autophagocytosis) 결정기(de- terminant)인 Atg16L1을 세포막(plasma membrane)으로 보충함으로써, autophagy의 기능조절에 관여한다.33 Auto- phagy라 함은 lysosome을 이용하여 세포 자신의 구성 성분 을 파괴하는 작용을 말한다. 따라서 autophagy가 작동되어 세포가 파괴되면, 세포 내부에 있는 각종 인자들이 밖으로 유 출된다. 이렇게 유출된 DAMPs 자체가 염증반응을 촉발시킬 수도 있고, NLRs이 인지하여 염증반응을 촉발시키게 된다.34 이 autophagy 결정기 Atg16L1을 coding하고 있는 유전자의 polymorphisms이 크론병 발현에 관여하는 위험인자임이 밝 혀지고 있어서 autophagy가 IBD 발병에 관여될 가능성이 높
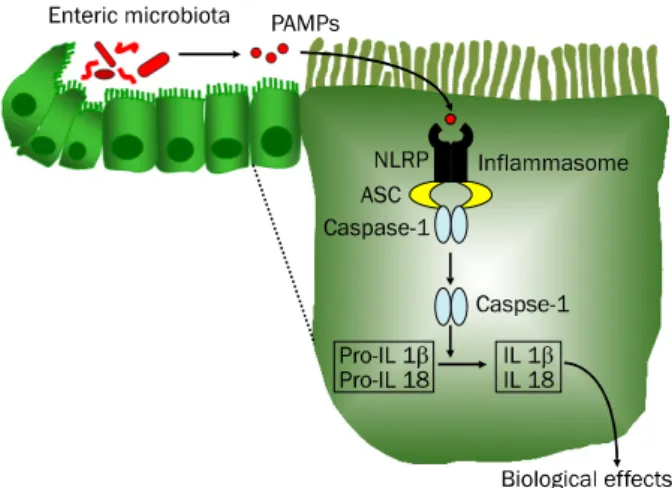
Fig. 1. Inflammasome signaling pathways. Nucleotide-binding oligo- merization domain-like receptor (NLRP) within the cytosol of intestinal epithelial cells recognizes pathogen-associated molecular patterns (PAMPs) of the enteric microbiota and forms a multiprotein complex with apoptosis-associated speck-like protein containing ‘a caspase recruitment domain (CARD)’ and caspase-1 called the
‘inflammasome.’ NLRP3 is capable of sensing both PAMPs and endogenous damage-associated molecular patterns (DAMPs). The inflammasome plays a central role in the inflammatory process by activating caspase-1 and mediating production of proinflammatory cytokine interleukin (IL)-1β and IL-18. In addition, caspase-1 activation mediates pyroptosis, a specific form of early cell death induced by intracellular pathogens that promotes cellular lysis and the release of intracellular inflammatory contents to stimulate additional inflammatory signaling pathways.
다.35-38 최근 IBD와 연관된 또 다른 NLR family member로
NLRP3에 대한 연구가 갑작스럽게 떠오르고 있다. 그러나 Nod1과 Nod2와는 달리, NLRP3의 활성은 inflammasome 이라는 세포질 내의 caspase-1-활성 복합체의 조립을 유도함 으로써 시작된다.
2. NLRP3 inflammasome의 구성
NLRP3는 chromosome 1q44에 위치한 NLRP3 유전자에 서 전사된 1,016개의 아미노산으로 되어있는 단백이다. 이 NLRP3는 면역세포와 IECs 모두에서 발현된다.39,40 NLRP3 와 더불어 apoptosis-associated speck-like protein con- taining ‘a caspase recruitment domain (CARD)’ (ASC)과 caspase-1이 ‘inflammasome’이라고 하는 multi-protein complex (분자량 700 kDa 이상)를 형성하고, 이 complex는 특정 조건이 되면 caspase-1의 활성을 촉발한다. Caspase-1 은 IL-1β ICE으로 알려진 protease로, 미성숙 cytokine인 pro-IL-1β와 pro-IL-18 등을 성숙한 형태로 만드는 역할을 한
다.20,41 즉, caspase-1이 활성화되면 전구물질(precursor) 형
태의 pro-IL-1β와 pro-IL-18 등이 활성화된 형태의 친염증성 cytokine으로 성숙되어 세포질 밖으로 분비됨으로써, 비로소
생물학적 활성을 나타내게 된다(Fig. 1). 또 한편으로는 cas- pase-1의 활성화가 pyroptosis라는 특이한 형태의 세포사멸 을 유도하기도 한다.42 이 pyroptosis는 병원균(intracellular pathogen)이 세포질 내부로 침입한 초기에 나타나는 반응으 로, 세포를 분해시켜 세포질 내부에 있는 각종 물질을 유출시 킴으로써 염증반응을 촉발시키는 특이한 세포반응이다.43,44 한편 지금까지 알려진 inflammasome 중에서 PAMPs와 DAMPs를 모두 인지할 수 있는 것은 NLRP3 inflammasome 이다. 따라서 pyroptosis에 의해 유출된 물질 DAMPs는 in- flammasome의 활성을 더욱 촉진시킬 수 있다. 또한 유출된 물질 중에는 염증반응을 조절하는 물질(예, prostaglandin 등)도 포함되어 있다는 점에서, NLRP3 inflammasome의 실 질적인 작동인자(effector)는 cytokine IL-1β와 IL18, 그리고 pyroptosis에 의해 유출되는 세포내 염증조절인자라고 할 수 있다. 그러나 최근 pro-IL-1β와 pro-IL-18 등을 성숙한 형태 로 만드는 과정이 inflammasome과는 별개로 진행된다는 보
고45,46가 있음을 고려해 볼 때, 오늘날 inflammasome을 통해
나타날 것으로 여겨지는 생체 반응들이 inflammasome과는 무관한 현상일 가능성도 있음을 밝히고자 한다.
3. NLRP3 inflammasome이 IBD 발병에 관여되어 있을 가 능성
IBD의 병인론을 규명하기 위해서는 이를 증명하기 위한 동 물모델이 필요하다. 그러나 현재까지 사람의 IBD에서 관찰되 는 임상적 특징을 모두 반영할 수 있는 완벽한 대리 모델은 개발되어 있지 않다. 따라서 실험적으로 중요한 몇가지 특징 만을 나타내는 장염 마우스 모델을 이용하고 있다.47 대표적으 로 dextran sodium sulfate (DSS) 모델은 대장염의 면역병리 기전을 연구하는데 광범위하게 사용되고 있다. DSS를 구강 투여하면 IECs에 독성을 나타낼 뿐만 아니라, 장관 내부에만 존재해야 할 장내 균무리의 위치가 파괴됨으로써 염증반응을 유발시킨다.6 DSS 모델의 임상적 특징은 체중감소, 설사, 직 장 출혈을 유도하고 사망에까지 이르게 한다. 또한 조직병리 분석결과 crypt와 IECs의 광범위한 손상, 호중구와 대식세포 의 현저한 침윤, 궤양 등과 같은 병리기전이 관찰된다.48
그러나 IBD 질환의 중증도는 IECs 뿐만 아니라 T 세포도 관여하는데, 이 모두를 반영할 수 없다는 점이 이 모델의 단점 이라고 할 수 있다. 즉, DSS 모델이 T 세포보다는 주로 급성 IECs 손상을 반영한다는 점이다. 그렇지만 T 세포가 결핍된 동물에서도 DSS에 의해 장염이 유발된다는 점,49-51 그리고 다 른 모델에 비해 DSS 모델이 손상된 IECs와 장벽기능 회복 및 악화를 쉽게 확인할 수 있을 뿐만 아니라 IBD 발현과 질병 조절에 관여하는 면역반응을 측정하는데 용이하다는 점에서 오늘날 가장 흔하게 사용되는 모델이다.
DSS 실험모델을 이용한 여러 연구들은 NLRP3 inflamma- some에서 유도되는 IL-1β와 IL-18의 생산이 장염의 원인이 될 수 있음을 보여주고 있다. 즉, NF-κB에 의해 발현이 조절 되는 IL-1은 염증반응을 유발하는 대표적인 cytokine이다. 또 한 IL-1 자체가 NF-κB를 활성화시킬 수 있는 강력한 인자이 기도 하다. 따라서 IL-1은 결국 NF-κB의 활성을 증폭시켜, 이에 의해 조절되는 각종 친염증성 매개체의 발현이 증가하여 과도한 염증반응을 유도하게 된다.52,53 이를 뒷받침하는 근거 로서 IBD 질환의 중증도가 IL-1의 발현 정도와 비례할 뿐만 아니라,54 IL-1을 억제할 경우 증상이 완화된다는 보고를55 들 수 있다. 그런데 IL-1 수용체가 결핍된 마우스(IL1r-/- mice)에 DSS로 장염을 유발시키면 정상마우스보다 병리조직 변화가 더 심하게 나타난다는 실험결과가 발표되었다.56 이 결과는 IL-1이 단순히 IBD의 염증반응 증폭에만 관여할 것이라는 기 존의 이론에 논란을 불러일으키게 되었다.
한편 IL-18은 IBD 환자의 장점막 조직과 혈액에서의 농도 가 정상인보다 증가되어 있다.57-59 또한 IL-18이 IBD 발현의 중요한 인자임을 제시한 논문들도 많이 발표되고 있다.60-63 특 히 동물실험모델에서 IL-8에 대한 항체 또는 이의 억제물질을 투여할 경우 질병이 완화됨이 관찰된다.61,62,64-66 따라서 IL-18 과 이의 억제물질 사이의 균형 파괴가 IBD 발현에 관여할 가 능성이 제시되고 있다.57 이와 같은 이유 때문에 이들 cyto- kine에 대한 억제물질을 IBD 치료제로 개발하려는 움직임이 활발하다.52 흥미롭게도 NLRP3-/- 마우스에서 DSS로 장염을 유도할 경우, 정상 마우스에 비해 증상이 완화되었다는 최근
보고는28,64 NLRP3 inflammasome이 IBD 발병 및 진행에 관
여하는 인자라는 점을 제시해 주고 있다.
이와 같이 NLRP3 inflammasome 신호전달 경로 하부에 위치하는 IL-1β와 IL-18이 IBD 증상 발현과 밀접한 관련이 있다는 보고들이 있는 반면, 오히려 IBD에 대한 방어인자로 작용할 것이라는 가설을 제기하는 논문들도 발표되고 있다.
이런 최근의 관찰을 기본으로, 이번 종설의 다음 단계에서는 IL-18에 초점을 맞추어 방어인자로서의 역할에 대해 기술하 고자 한다.
4. NLRP3 Inflammasome이 IBD 발병의 억제인자로 작용할 가능성
1) Inflammasome 실행 유전자가 IBD의 위험 대립유전자 (risk allele)일 가능성
크론병 환자의 골수세포(myeloid cell)를 세균 세포벽의 peptidoglycan 구성성분인 muramyl dipeptide (MDP)로 자 극하면 inflammasome의 작동인자인 IL-1β의 분비가 감소된 다.67-69 또한 IL-18과 IL-18 수용체 보조단백(receptor acces- sory protein)을 coding하고 있는 유전자의 polymorphism은
크론병에 대한 민감성의 증가와 관련이 있다.70,71 이런 보고들 은 IBD에서 inflammasomes이 중요한 역할을 수행할 가능성 을 제시해 준다. 또한 NLRP3 regulatory elements의 SNP가 크론병 발현에 대한 감수성 증가와 관련되어 있다는 최근 보 고는 이런 가설을 지지해줄 수 있는 연구결과라고 할 수 있 다.22 그러나 IBD 병리기전이 IL-1β와 IL-18 분비 감소와 직 접적으로 연관되어 있을 거라는 가설은 분자생물학적으로 아 직까지 분명하게 밝혀지지 않았다. 또한 NLRP3과 크론병과 의 유전적 관련성을 부정하는 보고도72 있음에 비추어 IBD 병인론에 있어서 inflammasome 실행 유전자가 IBD의 위험 대립유전자일 가능성에 대해서는 아직까지 논란이 많다고 할 수 있다.
2) 장벽 유지를 위한 NLRP3 inflammasome의 역할 점막면역계(mucosal immune system)는 병원균의 감염 을 방어함과 동시에 장내 균무리가 숙주와 평화롭게 공존할 수 있도록 다양한 면역/생리반응을 조절하고 있다. 그런데 상 당수의 IBD 환자에서 장벽의 파괴가 관찰되는데, 이 장벽파괴 는 점막면역계의 기능 손상과 연관되어 있다. 한편 피부상처의 회복에 IL-18이 관여한다는 보고를73 고려해 볼 때, NLRP3 inflammasome의 실행인자인 IL-18이 IBD에 의한 점막손상 을 회복시킬 가능성이 있다. 이에 대한 연구결과를 소개한다.
NLRP3 inflammasome이 결여된 마우스(NLRP3-/- 마우스) 는 장벽기능이 손상되어 대장염에 잘 걸린다.74 또한 NLRP3 inflammasome이 결여된 장벽에서는 대장조직 내부로 장내 미생물무리의 자리옮김(translocation)이 증가하고, 그 주위 의 림프조직 뿐만 아니라 비장과 간으로의 균 전파가 증가된
다.74,75 한편 IECs가 손상되면 이들을 대체하기 위해 장점막
crypt의 기저부에 있는 stem cell의 분열이 증가하여 손상된 IECs를 복구하려는 회복반응(repair response)이 관찰된다.76 그런데 NLRP3-/- 마우스에 DSS로 대장염을 유발할 경우 IECs 의 증식이 감소된다.74,75 이와 같은 관찰들은 NLRP3 in- flammasome이 IECs로 분화되는 stem cell의 증식을 촉진 함으로써 장벽유지에 중요한 역할을 할 것이라는 점을 시사해 준다.
NLRP3 inflammasome은 IL-1β와 IL-18을 생성해 내는 인자인데, 이 중 IL-18이 장벽방어에 더 중요할 것으로 추정 하고 있다.74,75,77 그 이유는 DSS로 장염을 유발시키면 IL-18 의 생성은 상당히 많은 양의 증가를 보인 반면, IL-1β의 증가 는 미약하기 때문이었다. 또한 크론병 환자의 대장 IECs에서 IL-18의 발현 증가가 확인되고 있다.59,78 무엇보다 위의 가설 을 지지하는 증거로 IL-18은 IECs의 증식과 손상된 상피세포 의 회복반응에 관여한다는 보고를 들 수 있다.78,79 따라서 IECs의 NLRP3 inflammasome를 매개로 생성되는 IL-18은 장점막을 회복시키고 DSS에 의한 장염을 방어할 수 있는 매
Fig. 2. Hypothesis of nucleotide-binding oligomerization domain-like receptor (NLRP) 3 inflammasome-mediated protection against dextran sodium sulfate (DSS)-induced colitis and colitis-associated colon cancer (CAC) in mice model. DSS causes direct intestinal epithelial cell (IEC) injury, increasing permeability and translocation of bacteria into the mucosa, leading to inflammatory responses that include the recruitment of immune cells such as neutrophils and macrophages. In the stage of acute colitis, the production of interleukin (IL)-18 by the NLRP3 inflammasome may be involved in IEC repair. Chronic inflammation can lead to carcinogenesis via production of DNA-damaging oxygen radical species, proinflam- matory mediators that promote cellular survival, and increases of epithelial proliferation and angiogenesis. In a murine model, the carcinogen azoxymethane is used to introduce genomic mutations by methylation. Although the mechanism still remains unclear, the downstream signaling of IL-18 and interferon-γ may play a role in inhibiting epithelial proliferation.
개체라고 할 수 있다. 그러나 IL-18이 IECs의 증식과 회복을 어떻게 촉진시키는지에 대한 기전은 아직까지 확실하게 밝혀 지지 않고 있다.
3) 장관의 항상성(gut homeostasis) 유지를 위한 NLRP3 inflammasome의 역할
정상적인 면역반응이라 함은 비병원성 장내 미생물무리에 대해서는 면역관용을 유지하고 병원균에 대해서는 효과적인 방어면역이 작동되는 것을 의미한다. 이런 면역반응의 균형은 장관의 항상성을 유지하는데 대단히 중요하다. 그런데 IBD 환자에서는 이런 항상성이 파괴되어 있는 경우가 흔하다. 그 렇지만 IBD 진행 과정 중 급성염증반응이 나타나더라도 이를 극복할 수 있는, 즉 항상성을 회복시킬 수 있는 반응이 나타난 다. 이와 관련된 최근의 연구결과들을 소개한다.
앞서 기술한 바와 같이 NLRP3-/- 마우스는 DSS에 의한 장 염에 더 민감하게 반응한다.74,77,80 또한 이 마우스에 2,4,6-tri- nitrobenzene sulfonate (TNBS)를 투여해 급성대장염을 유 발시킬 경우, 체중 감소, 설사, 결장 출혈과 사망률이 일반 마 우스에 비해 더 심하게 나타난다.74,80 NLRP3-/- 마우스와 유사 하게, inflammasome 구성단백인 ASC와 caspase-1이 결핍 이 된 모델에서도 DSS에 의한 장염의 병리조직 변화와 사망 률이 더 높게 관찰된다.74,75,77 또한 NLRP3-/- casp1-/- 마우스 에 급성장염을 유발할 경우 IECs의 증식이 감소한다.74 그리 고 이 마우스에 DSS를 투여하여 장염을 유발할 경우, 장벽 투과성의 증가와 더불어 장내 미생물무리가 전신으로 확산되 는 것을 관찰할 수 있다.74,75 이런 결과들은 DSS가 IECs에 독성인자로 작동함에도 불구하고, NLRP3 inflammasome이 정상적으로 작동할 경우에는 DSS에 의한 반응을 회복시키는 데 필요한 각종 시그널을 촉발할 수 있을 것이라는 가설을 가능하게 해 준다(Fig. 2). 따라서 NLRP3 inflammasome 신 호전달 하부에 위치한 IL-1β와 IL-18이 IECs의 항상성 회복 에 관여할 것으로 보인다.
그런데 NLRP3-/- casp1-/- 마우스에 DSS로 장염을 유발시 킬 경우 정상 마우스보다 더 심한 증상이 나타나는데, 이때 비-조혈세포(non-hematopoietic cell)에서 NLRP374 또는 caspase-175의 기능을 회복시키면 그 증상이 상당히 완화된 다. 그리고 IBD 환자에서의 IL-18 생성은 고유층(lamina pro- pia)에 분포하는 세포에서도 관찰된다.59 따라서 IBD 질환 경 과에 있어서 IL-18의 근원은 IECs이 유일한 것은 아니라고 할 수 있다.
장벽의 투과성 유지와 더불어, IECs의 항상성을 유지시키 는 요소 중의 하나로 항-미생물 펩타이드(anti-microbial peptide)를 들 수 있다. 즉, 인체에서는 장관내 감염을 낮추고 미생물무리의 숫자를 일정하게 유지하기 위해, 장관에 분포하 는 IECs와 수지상세포 등이 장내 미생물무리와 접촉하여 각
종 항-미생물 펩타이드를 발현시킨다.81 미생물 증식 억제 기 능을 나타내는 물질로 defensin, cathelicidin LL-37, lyso- zyme, phospholipase A, 세균살해 기능을 갖는 각종 단백 (예, ubiquicidin, ribosomal protein, eosinophilic protein) 이 알려져 있다.82
이들 중에서 대표적인 항-미생물 펩타이드로 18-45개의 아 미노산(분자량 약 3.5-6 kDa)으로 구성된 펩타이드 defensin
이 잘 알려져 있다. Defensin은 세균 세포막과 결합하여, 세 포막에 구멍을 냄으로써 세균 세포를 파괴한다. 이 defensin 은 크게 α-defensin과 β-defensin으로 나눌 수 있다. 사람에 서의 α-defensin은 호중구에서 분비되는 4종류의 α-de- fensin (human neutrophil protein [HNP]-1, HNP-2, HNP-3 및 HNP-4)과 Paneth cell에서 분비되는 2종류의 α-defensin (human α-defensins [HD]-5 및 HD-6)이 보고되 어 있다.83 그리고 β-defensin은 장관에서 기본적으로 (constitutive) 발현되는 human β-defensin (hBD)-1과 병원 균 등의 자극에 의해 유도되는 hBD-2로 나눌 수 있다.82
NLRP3-/- 마우스의 대장 crypt는 항-미생물 활성이 감소된 양상을 보이는데, 이 현상은 대장의 defensin 발현 변화와 연 관되어 있다.80 이런 동물실험 결과는 크론병 환자에서 de- fensin의 발현이 감소되어 있다는 점에서84,85 인체에서도 동 일한 현상으로 나타날 가능성이 높다. 그러나 defensin 발현 이 NLRP3 inflammasome에 의해 직접적으로 조절되는지 혹 은 IL-1β 또는 IL-18에 의해 나타나는 생물학적 신호전달 하 부에 해당되는 현상인지는 아직까지 확실하지 않다. 그럼에도 불구하고 NLRP3-/- 마우스에서의 대장 defensin 발현 감소는 IBD 방어에 참여하는 NLRP3 inflammasome의 역할을 시사 해 준다고 할 수 있다.
4) NLRP3 Inflammasome 작동인자 IL-1β와 IL-18이 IBD 병인에 미치는 영향
앞에서 NLRP3 inflammasome이 정상적인 장벽 유지에 중요한 역할을 한다는 이론을 제시한 바 있다. 즉, IECs가 손 상을 받더라도 이를 회복시키기 위해 inflammasome 작동인 자인 IL-1β와 IL-18가 관여할 가능성이 있다.78 Inflammaso- me을 매개로 이들 cytokine이 분비되면, IECs와 점막 내부 의 면역세포 표면에 존재하는 수용체와 결합하여 생물학적 기 능을 발휘한다. 그런데 IL-1 수용체 결핍 마우스(IL1r-/- mice) 에 DSS로 장염을 유발시킬 경우 정상마우스보다 병리조직 변 화가 더 심하게 관찰된다.56 또한 IL18-/- IL18r1-/- 마우스에 DSS로 장염을 유발시키면, 정상 마우스보다 더 심한 병리조 직 변화와 더 높은 사망률이 관찰된다.79 한편 DSS에 의한 장염은 IL-1β와 IL-18의 분비에 필수적으로 요구되는 adap- tor protein MyD88이 결여된 마우스에서 더 심하게 나타난 다.6,86,87 따라서 앞서 기술한 NLRP3-/- casp1-/- 마우스에서 DSS-유도 장염이 더 심하게 관찰되는 이유가 IL-1β와 IL-18 의 분비 장애로 인해 초래될 것이라는 가능성이 제시된다. 이 이론은 DSS로 장염을 유발시킨 casp1-/- 마우스에 재조합 IL-18를 외부에서 투여할 경우 그 증상이 완화된다는 보고에 의해 그 가능성이 더욱 높아진다고 할 수 있다.74,75 따라서 IBD 발생/진행과정에 있어서 NLRP3 inflammasome이 방어 기전으로 작동한다는 이론은 이의 작동인자인 IL-1β와 IL-18
에 의해 수행된다고 할 수 있다.
5) NLRP3 inflammasome 역할 규명을 위해 사용한 특정유 전자 제거모델과 생화학적 접근모델 간의 결과의 불일치 앞서 기술한 특정유전자를 제거한 마우스 모델에서의 결과 들을 caspase-1과 IL-18을 중화시킨 생화학적 접근모델의 결 과들과 비교해 보면, 그 결과들이 서로 일치하지 않는 경우가 많다. 즉, 앞서 IL18-/- IL18r1-/- 마우스 또는 casp1-/- 마우스 모델을 이용하여 IL-18이 IBD 병인에 방어적으로 작용할 가 능성을 제시한 연구결과들을 소개하였다.74,75,77,79,80
그런데 재 조합 IL-18 결합단백 또는 IL-18 항체를 이용한 연구에서 IL-18이 장염발생을 촉진한다는 보고들이 있다.65,66 또한 DSS-유발 장염모델에 caspase-1의 화학적 억제제인 pralna- casan을 투여하면 장염이 완화된다는 결과로 미루어 cas- pase-1은 IBD 발생을 촉진하는 인자일 가능성이 제시된
다.64,88,89 따라서 NLRP3 inflammasome 작동인자들이 IBD
의 병인론에서 방어인자로 작용할 가능성이 있다는 이론은 논 란의 여지가 있다고 할 수 있다.
일반적으로 표적유전자 제거(gene-targeted deletion)를 이용한 동물모델은 특정 단백의 활성을 차단하는 확실한 방법 이라고 알려져 있기 때문에 생화학적 방법보다 우월한 접근법 이라고 생각하는 경향이 있다. 그러나 IBD 환자에 대한 치료 적 접근은 유전자조작 방법보다는 화학적 방법이 더 현실적이 라는 점에서 생화학적 접근모델의 결과를 무시할 수 없다는 점을 강조하고자 한다.
6) 장염과 연관된 대장암(colitis-associated colon cancer, CAC)에 있어서의 NLRP3 inflammasome의 역할 일반적으로 염증반응은 상처와 감염에 대한 방어를 목표로 하는 숙주반응으로 생각하고 있다. 그렇지만 만성염증은 DNA 에 손상을 줄 수 있는 oxygen radical species 뿐만 아니라 상피세포의 생존과 증식, 그리고 혈관형성(angiogenesis)을 촉진시킬 수 있는 다양한 친염증성 매개체의 분비를 통해 종 양발생을 촉진시킨다.42,90 따라서 IBD에서는 장점막 염증반응 이 오래 지속되면 결국 장암으로 발전할 수 있는 위험 인자로 간주되고 있다.91
IBD와 관련된 종양발생 모델로 DSS와 azoxymethane (AOM)을 투여한 마우스가 주로 사용되고 있다. 즉, DSS로 IBD와 유사한 장염을 발생시킴과 동시에, 발암물질(carcino- gen)인 AOM은 상피세포의 메틸화를 통해 유전자 변이를 유 도하여 종양을 발생시킨다.92,93 이 모델은 단점이 있음에도 불 구하고 장염과 연관된 종양발생 시그널들을 확인하는 데 가장 흔하게 이용되고 있다.42
AOM/DSS 모델을 이용한 연구들을 종합해 보면, NLR 활 성 결여는 장점막 세포증식 및 종양발생 가능성의 증가와 일 치하는 경향을 보여주고 있다. 예를 들어, Nod1 활성화 경로
를 제거한 상태에서 장염을 유발시킬 경우, IEC 층의 투과성 을 더욱 악화시키고 CAC 발생을 촉진하였다.94 또한 Nod2 polymorphism은 장관 종양의 발생 증가와 연관되어 있다는 연구결과도 있다.95 현재까지 CAC 발생에 NLRP3가 직접적 으로 관여한다는 연구결과는 없다. 그러나 NLRP3-/- 마우스를 AOM/DSS로 처리했을 때 염증반응이 증가하고 장벽 붕괴의 결과로 dysplasia와 CAC 발생이 증가했다.77,96 또한 NLRP3 inflammasome의 구성인자인 ASC와 caspase-1을 선택적으 로 제거한 마우스에서도 비슷한 결과들이 관찰된다.77,96 이와 같은 결과들은 NLRP3 inflammasome이 IBD 진행 과정에 있어서의 CAC 발생에 억제인자로 작용할 가능성을 시사해 준다.
NLRP3 inflammasome이 CAC 발생을 억제할 수 있는 기 전으로 IL-18이 거론되고 있다. 즉, IL-18은 실험 종양모델에 서의 육종(sarcoma)과 흑색종(melanoma)에 대해 항-종양 효 과를 나타낸다.95,97,98 또한 IL-18은 종양성장과 혈관형성을 억
제하고,99-101 손상된 상피세포층의 회복을 촉진시킨다.75,78,96
그리고 DSS를 투여한 NLRP3-/- casp1-/- 마우스의 대장과74,75 AOM/DSS를 처리한 NLRP3-/- casp1-/- 마우스의 대장에서77,96 IL-18의 발현이 유의하게 감소하였다. 한편 AOM/DSS를 처 리한 casp1-/- 마우스에 재조합 IL-18을 투여하면, 질병 진행 이 현저하게 억제되었다.96 또한 AOM/DSS를 처리한 IL18-/- IL18r1-/- 마우스는 정상 마우스보다 장폴립(intestinal polyp) 의 숫자가 증가되었다.102 이와 같은 결과들은 CAC 발생을 억제할 수 있는 기전으로써, NLRP3 inflammasome 신호전 달계 하부에 위치한 IL-18이 중요하다는 점을 제시해 준다.
그러나 장점막의 NLRC4 inflammasome도 장염과 연관된 종양발생을 억제할 수 있다는 보고가 있음에 비추어,103 CAC 발생 억제에 NLRP3 inflammasome 뿐만 아니라 다른 인자 도 관여할 가능성이 있다. 그러므로 NLRP3 또는 NLRC4 를 coding하는 유전자에 결함이 있는 개체는 IBD와 CAC 발생 에 대한 감수성이 높아질 것으로 추정된다.
7) IBD 진행 단계에 따른 NLRP3 inflammasome 실행인자 IL-18의 이중적 역할
앞서 IL-18은 손상된 IECs을 회복할 수 있도록 장세포 증 식(enterocyte proliferation)을 촉진함으로써 IBD의 방어인 자로 작용할 것이라는 이론을 제시한 바 있다. 따라서 CAC 병인론에 있어서 IL-18이 종양세포의 증식을 억제한다는 가 설은 앞서 기술한 이론과 모순된다고 할 수 있다. 즉, IBD 급 성기와 만성기에서의 IL-18 역할이 서로 일치하지 않는다는 점이 이 가설의 단점이라고 할 수 있다. 이 모순을 해결하기 위하여, 염증 진행시기에 따라 IL-18의 기능이 달라진다는 가 설을 제시하고 있다.104 마지막으로 이 가설의 타당성을 기술 하고자 한다.
장염의 급성기에 IL-18은 crypt 기저부에서 stem cell의 증식을 통해 손상된 IECs의 교체를 유도함으로써 장벽의 기 능을 회복할 수 있도록 작용할 것으로 보인다. 이렇게 함으로 써 장내 미생물무리의 전신적 확산과 과도한 염증반응을 방지 하게 된다. 그러나 이 시기에서 회복되거나 만성화 과정에 도 달하면 IL-18은 장벽의 종양부위에서 IECs 증식을 억제할 것 으로 보인다(Fig. 2). 이와 같은 추정은 IL-18이 interferon (IFN)-γ 발현을 촉진시키는 Th1 세포반응을 유도할 뿐만 아 니라 자연살해세포(natural killer cell)의 숫자를 증가시킨다 는 이론에 근거한다.105 또한 AOM/DSS를 처리한 NLRP3-/- casp1-/- 마우스의 대장에서 INF-γ의 발현이 현저하게 감소되 었다는 보고는 IL-18과 IFN-γ가 CAC 발생과정 중 방어역할 을 할 것이라는 가설을 가능하게 해 준다.96 IFN-γ는 IFN-γ 수용체(IFN-γR)를 매개로 전사인자인 STAT1을 핵 내부로 이 동(nuclear translocation)시켜 생물학적 활성을 나타낸다.106 이 생물학적 활성 중의 하나로 종양세포 증식억제가 잘 알려 져 있다.107 그런데 AOM/DSS를 처리한 casp1-/- 마우스의 대 장에서는 활성화된 STAT1 level이 현저하게 감소하는데, 이 때 외부에서 IFN-γ 또는 IL-18을 추가해 주면 이런 현상이 완화된다.104
IL-18의 이중적 역할을 뒷받침할 수 있는 또 다른 증거로, DSS로 유도한 장염 모델에서 IFN-γ는 질병 초기단계에서 IECs의 증식을 촉진하지만, 후기단계에서는 증식억제로 작용 할 수 있을 것이라는 보고를 들 수 있다.108 그러나 IFN-γ가 IEC 층의 tight junction을 파괴하고 장세포 이동(IEC mi- gration)을 억제함으로써 IBD 발병에 기여한다는 논문들도 보고되고 있다는 점에서,109-112 이 가설에 대한 논란은 불가피 한 것으로 보인다. 앞으로 이 가설에 대한 검증과정이 활발해 질 것으로 예상되며, 이를 통해 IBD 병인론에 대한 지식이 한층 발전될 것으로 보인다.
결 론
최근 궤양성 대장염과 크론병을 대상으로 NLR 단백인 Nod1과 Nod2의 역할이 규명됨에 따라 IBD 발병기전이 보다 구체적으로 정립되고 있다. 이와 더불어 NLRP3 inflamma- some이 IBD 발현에 방어적으로 작용할 것이라는 연구결과는 IBD 병인론에 관한 지식을 한 단계 더 끌어올렸다고 할 수 있다. 즉, inflammasome 신호전달경로 하부에 위치하는 각 종 생물학적 효과들이 IBD 발병에 기여할 것이라는 학설이 아직까지 대세를 이루고 있음에도 불구하고, IL-18에 의한 반 대 이론이 갑작스럽게 부상하고 있다. 이번 종설은 NLRP3 inflammasome이 IBD 병인론에 있어서 질병 발현을 촉진하 는 역할을 먼저 기술한 뒤, 이에 대한 반론으로 방어역할을
할 수 있음을 증명한 결과들을 가능한 객관적으로 기술하였 다. 그러나 최근 pro-IL-1β와 pro-IL-18 등을 성숙한 형태로 만드는 과정이 inflammasome과는 별개로 진행된다는 연구 결과들도 발표되고 있어서, inflammasome을 통해 나타날 것 으로 여겨지는 생체 반응들이 이와는 무관한 현상일 가능성도 있다. 또한 동일한 실험모델에서 반대의 결과들을 보고한 논 문들도 있음에 비추어, IBD에서의 NLRP3 inflammasome의 정확한 역할은 아직까지 논쟁의 여지가 있다는 점을 지적하고 자 한다. 앞으로 IBD 발생과 진행과정에 관여하는 NLR과 in- flammasome의 복잡한 역할들이 점차 밝혀질 것으로 보인 다. 이러한 지식의 발전이 IBD와 이와 연관된 종양에 대해 새로운 치료 전략을 제시해 줄 것으로 기대한다.
REFERENCES
1. Cario E. Toll-like receptors in inflammatory bowel diseases: a decade later. Inflamm Bowel Dis 2010;16:1583-1597.
2. Wells JM, Rossi O, Meijerink M, van Baarlen P. Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci U S A 2011;108(Suppl 1):4607-4614.
3. Koh YS. Nucleic acid recognition and signaling by Toll-like re- ceptor 9: compartment-dependent regulation. J Bacteriol Virol 2011;41:131-132.
4. Chen GY, Nuñez G. Sterile inflammation: sensing and react- ing to damage. Nat Rev Immunol 2010;10:826-837.
5. Abreu MT. Toll-like receptor signalling in the intestinal epi- thelium: how bacterial recognition shapes intestinal func- tion. Nat Rev Immunol 2010;10:131-144.
6. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004;118:229-241.
7. Bouskra D, Brézillon C, Bérard M, et al. Lymphoid tissue gen- esis induced by commensals through NOD1 regulates in- testinal homeostasis. Nature 2008;456:507-510.
8. Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol 2011;11:9-20.
9. Kang JS. Journal walk regarding the expanding role of micro- biota in our gut. J Bacteriol Virol 2011;41:63-64.
10. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol 2010;28:573-621.
11. Sartor RB. Microbial influences in inflammatory bowel disea- ses. Gastroenterology 2008;134:577-594.
12. Kim JM. Inflammatory bowel diseases and enteric microbiota.
Korean J Gastroenterol 2010;55:4-18.
13. Abraham C, Medzhitov R. Interactions between the host in- nate immune system and microbes in inflammatory bowel disease. Gastroenterology 2011;140:1729-1737.
14. Manichanh C, Rigottier-Gois L, Bonnaud E, et al. Reduced di- versity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut 2006;55:205-221.
15. Kleessen B, Kroesen AJ, Buhr HJ, Blaut M. Mucosal and invad- ing bacteria in patients with inflammatory bowel disease compared with controls. Scand J Gastroenterol 2002;37:
1034-1041.
16. Goyette P, Labbé C, Trinh TT, Xavier RJ, Rioux JD. Molecular pathogenesis of inflammatory bowel disease: genotypes, phenotypes and personalized medicine. Ann Med 2007;39:
177-199.
17. Kanneganti TD, Lamkanfi M, Núñez G. Intracellular NOD-like receptors in host defense and disease. Immunity 2007;27:
549-559.
18. Werts C, Rubino S, Ling A, Girardin SE, Philpott DJ. Nod-like receptors in intestinal homeostasis, inflammation, and can- cer. J Leukoc Biol 2011;90:471-482.
19. Martinon F, Burns K, Tschopp J. The inflammasome: a molec- ular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002;10:417-426.
20. Hong S, Park S, Yu JW. Pyrin domain (PYD)-containing in- flammasome in innate immunity. J Bacteriol Virol 2011;41:
133-146.
21. Hornung V, Ablasser A, Charrel-Dennis M, et al. AIM2 recog- nizes cytosolic dsDNA and forms a caspase-1-activating in- flammasome with ASC. Nature 2009;458:514-518.
22. Villani AC, Lemire M, Fortin G, et al. Common variants in the NLRP3 region contribute to Crohn's disease susceptibility.
Nat Genet 2009;41:71-76.
23. Kanneganti TD, Ozören N, Body-Malapel M, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 2006;440:233-236.
24. Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006;440:228-232.
25. Sutterwala FS, Ogura Y, Szczepanik M, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 2006;24:317- 327.
26. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009;27:519- 550.
27. Bauer C, Duewell P, Mayer C, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010;59:1192-1199.
28. Elinav E, Strowig T, Kau AL, et al. NLRP6 inflammasome regu- lates colonic microbial ecology and risk for colitis. Cell 2011;
145:745-757.
29. Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 2001;411:599-603.
30. McGovern DP, Hysi P, Ahmad T, et al. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum Mol Genet 2005;14:1245-1250.
31. Ng SC, Tsoi KK, Kamm MA, et al. Genetics of inflammatory bowel disease in Asia: Systematic review and meta-analysis.
Inflamm Bowel Dis 2011.
32. Rioux JD, Abbas AK. Paths to understanding the genetic basis
of autoimmune disease. Nature 2005;435:584-589.
33. Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma mem- brane at the site of bacterial entry. Nat Immunol 2010;11:
55-62.
34. Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011;474:298-306.
35. Henderson P, van Limbergen JE, Wilson DC, Satsangi J, Russell RK. Genetics of childhood-onset inflammatory bowel disease. Inflamm Bowel Dis 2011;17:346-361.
36. Hampe J, Franke A, Rosenstiel P, et al. A genome-wide associ- ation scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 2007;39:
207-211.
37. Lees CW, Barrett JC, Parkes M, Satsangi J. New IBD genetics:
common pathways with other diseases. Gut 2011;60:1739- 1753.
38. Stappenbeck TS, Rioux JD, Mizoguchi A, et al. Crohn disease:
a current perspective on genetics, autophagy and immunity.
Autophagy 2011;7:355-374.
39. Kummer JA, Broekhuizen R, Everett H, et al. Inflammasome components NALP 1 and 3 show distinct but separate ex- pression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem 2007;55:443-452.
40. Leemans JC, Cassel SL, Sutterwala FS. Sensing damage by the NLRP3 inflammasome. Immunol Rev 2011;243:152- 162.
41. Lamkanfi M, Dixit VM. The inflammasomes. PLoS Pathog 2009;5:e1000510.
42. Chen GY, Núñez G. Inflammasomes in intestinal inflam- mation and cancer. Gastroenterology 2011;141:1986-1919.
43. Fernandes-Alnemri T, Wu J, Yu JW, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating in- flammatory cell death via caspase-1 activation. Cell Death Differ 2007;14:1590-1604.
44. Fink SL, Cookson BT. Pyroptosis and host cell death re- sponses during Salmonella infection. Cell Microbiol 2007;9:
2562-2570.
45. Mayer-Barber KD, Barber DL, Shenderov K, et al. Caspase-1 independent IL-1beta production is critical for host resist- ance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 2010;184:3326-3330.
46. van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA.
Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol 2011;32:110-116.
47. Pizarro TT, Arseneau KO, Bamias G, Cominelli F. Mouse mod- els for the study of Crohn's disease. Trends Mol Med 2003;9:218-222.
48. Kim JM, Kang HW, Cha MY, et al. Novel guggulsterone de- rivative GG-52 inhibits NF-kappaB signaling in intestinal epi- thelial cells and attenuates acute murine colitis. Lab Invest 2010;90:1004-1015.
49. Axelsson LG, Landström E, Goldschmidt TJ, Grönberg A, Bylund-Fellenius AC. Dextran sulfate sodium (DSS) induced experimental colitis in immunodeficient mice: effects in
CD4(+) -cell depleted, athymic and NK-cell depleted SCID mice. Inflamm Res 1996;45:181-191.
50. Chen Y, Chou K, Fuchs E, Havran WL, Boismenu R. Protection of the intestinal mucosa by intraepithelial gamma delta T cells. Proc Natl Acad Sci U S A 2002;99:14338-14343.
51. Shintani N, Nakajima T, Okamoto T, Kondo T, Nakamura N, Mayumi T. Involvement of CD4+ T cells in the development of dextran sulfate sodium-induced experimental colitis and suppressive effect of IgG on their action. Gen Pharmacol 1998;31:477-481.
52. Hanai H, Takeda Y, Eberhardson M, et al. The mode of actions of the Adacolumn therapeutic leucocytapheresis in patients with inflammatory bowel disease: a concise review. Clin Exp Immunol 2011;163:50-58.
53. Tilg H, Moschen AR, Kaser A, Pines A, Dotan I. Gut, in- flammation and osteoporosis: basic and clinical concepts.
Gut 2008;57:684-694.
54. Dijkstra G, Moshage H, Jansen PL. Blockade of NF-kappaB activation and donation of nitric oxide: new treatment options in inflammatory bowel disease? Scand J Gastroenterol Suppl 2002;(236):37-41.
55. Siegmund B. Interleukin-1beta converting enzyme (caspase- 1) in intestinal inflammation. Biochem Pharmacol 2002;64:
1-8.
56. Lebeis SL, Powell KR, Merlin D, Sherman MA, Kalman D.
Interleukin-1 receptor signaling protects mice from lethal in- testinal damage caused by the attaching and effacing patho- gen Citrobacter rodentium. Infect Immun 2009;77:604-614.
57. Leach ST, Messina I, Lemberg DA, Novick D, Rubenstein M, Day AS. Local and systemic interleukin-18 and inter- leukin-18-binding protein in children with inflammatory bow- el disease. Inflamm Bowel Dis 2008;14:68-74.
58. Pagès F, Lazar V, Berger A, et al. Analysis of interleukin-18, in- terleukin-1 converting enzyme (ICE) and interleukin-18-re- lated cytokines in Crohn's disease lesions. Eur Cytokine Netw 2001;12:97-104.
59. Pizarro TT, Michie MH, Bentz M, et al. IL-18, a novel immunor- egulatory cytokine, is up-regulated in Crohn's disease: ex- pression and localization in intestinal mucosal cells. J Immunol 1999;162:6829-6835.
60. Kanai T, Watanabe M, Okazawa A, et al. Macrophage-derived IL-18-mediated intestinal inflammation in the murine model of Crohn's disease. Gastroenterology 2001;121:875-888.
61. Nakamura S, Otani T, Ijiri Y, Motoda R, Kurimoto M, Orita K.
IFN-gamma-dependent and -independent mechanisms in adverse effects caused by concomitant administration of IL-18 and IL-12. J Immunol 2000;164:3330-3336.
62. Ten Hove T, Corbaz A, Amitai H, et al. Blockade of endogenous IL-18 ameliorates TNBS-induced colitis by decreasing local TNF-alpha production in mice. Gastroenterology 2001;121:
1372-1379.
63. Wirtz S, Becker C, Blumberg R, Galle PR, Neurath MF.
Treatment of T cell-dependent experimental colitis in SCID mice by local administration of an adenovirus expressing IL-18 antisense mRNA. J Immunol 2002;168:411-420.
64. Bauer C, Loher F, Dauer M, et al. The ICE inhibitor pralnacasan
prevents DSS-induced colitis in C57BL/6 mice and sup- presses IP-10 mRNA but not TNF-alpha mRNA expression.
Dig Dis Sci 2007;52:1642-1652.
65. Siegmund B, Fantuzzi G, Rieder F, et al. Neutralization of inter- leukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am J Physiol Regul Integr Comp Physiol 2001;281:R1264-R1273.
66. Sivakumar PV, Westrich GM, Kanaly S, et al. Interleukin 18 is a primary mediator of the inflammation associated with dex- tran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut 2002;50:812-820.
67. Li J, Moran T, Swanson E, et al. Regulation of IL-8 and IL-1beta expression in Crohn's disease associated NOD2/CARD15 mutations. Hum Mol Genet 2004;13:1715-1725.
68. van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, et al.
Stimulation of the intracellular bacterial sensor NOD2 pro- grams dendritic cells to promote interleukin-17 production in human memory T cells. Immunity 2007;27:660-669.
69. van Heel DA, Ghosh S, Butler M, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn's disease. Lancet 2005;365:1794-1796.
70. Tamura K, Fukuda Y, Sashio H, et al. IL18 polymorphism is as- sociated with an increased risk of Crohn's disease. J Gastroenterol 2002;37(Suppl 14):111-116.
71. Zhernakova A, Festen EM, Franke L, et al. Genetic analysis of innate immunity in Crohn's disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am J Hum Genet 2008;82:1202-1210.
72. Lewis GJ, Massey DC, Zhang H, et al. Genetic association be- tween NLRP3 variants and Crohn's disease does not repli- cate in a large UK panel. Inflamm Bowel Dis 2011;17:1387- 1391.
73. Kämpfer H, Paulukat J, Mühl H, Wetzler C, Pfeilschifter J, Frank S. Lack of interferon-gamma production despite the presence of interleukin-18 during cutaneous wound healing.
Mol Med 2000;6:1016-1027.
74. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010;32:379-391.
75. Dupaul-Chicoine J, Yeretssian G, Doiron K, et al. Control of in- testinal homeostasis, colitis, and colitis-associated color- ectal cancer by the inflammatory caspases. Immunity 2010;
32:367-378.
76. Radtke F, Clevers H. Self-renewal and cancer of the gut: two sides of a coin. Science 2005;307:1904-1909.
77. Allen IC, TeKippe EM, Woodford RM, et al. The NLRP3 in- flammasome functions as a negative regulator of tumori- genesis during colitis-associated cancer. J Exp Med 2010;
207:1045-1056.
78. Reuter BK, Pizarro TT. Commentary: the role of the IL-18 sys- tem and other members of the IL-1R/TLR superfamily in in- nate mucosal immunity and the pathogenesis of in- flammatory bowel disease: friend or foe? Eur J Immunol 2004;34:2347-2355.
79. Takagi H, Kanai T, Okazawa A, et al. Contrasting action of IL-12
and IL-18 in the development of dextran sodium sulphate col- itis in mice. Scand J Gastroenterol 2003;38:837-844.
80. Hirota SA, Ng J, Lueng A, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis 2011;17:1359-1372.
81. Nuding S, Zabel LT, Enders C, et al. Antibacterial activity of hu- man defensins on anaerobic intestinal bacterial species: a major role of HBD-3. Microbes Infect 2009;11:384-393.
82. Yoon YM, Lee JY, Yoo D, et al. Bacteroides fragilis enterotoxin induces human beta-defensin-2 expression in intestinal epi- thelial cells via a mitogen-activated protein kinase/I kappaB kinase/NF-kappaB-dependent pathway. Infect Immun 2010;
78:2024-2033.
83. De Smet K, Contreras R. Human antimicrobial peptides: de- fensins, cathelicidins and histatins. Biotechnol Lett 2005;27:
1337-1317.
84. Simms LA, Doecke JD, Walsh MD, et al. Reduced alpha-de- fensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn's disease. Gut 2008;57:
903-910.
85. Wehkamp J, Harder J, Weichenthal M, et al. NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal alpha-defensin expression. Gut 2004;53:1658- 1664.
86. Araki A, Kanai T, Ishikura T, et al. MyD88-deficient mice devel- op severe intestinal inflammation in dextran sodium sulfate colitis. J Gastroenterol 2005;40:16-23.
87. Fukata M, Michelsen KS, Eri R, et al. Toll-like receptor-4 is re- quired for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol 2005;288:G1055- G1065.
88. Loher F, Bauer C, Landauer N, et al. The interleukin-1 be- ta-converting enzyme inhibitor pralnacasan reduces dextran sulfate sodium-induced murine colitis and T helper 1 T-cell activation. J Pharmacol Exp Ther 2004;308:583-590.
89. Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation.
Proc Natl Acad Sci U S A 2001;98:13249-13254.
90. Coussens LM, Werb Z. Inflammation and cancer. Nature 2002;420:860-867.
91. Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflam- mation. Am J Physiol Gastrointest Liver Physiol 2004;287:
G7-17.
92. Suzuki R, Kohno H, Sugie S, Tanaka T. Sequential ob- servations on the occurrence of preneoplastic and neo- plastic lesions in mouse colon treated with azoxymethane and dextran sodium sulfate. Cancer Sci 2004;95:721-727.
93. Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A nov- el inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate.
Cancer Sci 2003;94:965-973.
94. Chen GY, Shaw MH, Redondo G, Núñez G. The innate immune receptor Nod1 protects the intestine from inflammation-in- duced tumorigenesis. Cancer Res 2008;68:10060-10067.
95. Micallef MJ, Tanimoto T, Kohno K, Ikeda M, Kurimoto M.
Interleukin 18 induces the sequential activation of natural killer cells and cytotoxic T lymphocytes to protect syngeneic mice from transplantation with Meth A sarcoma. Cancer Res 1997;57:4557-4563.
96. Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti TD. IL-18 production downstream of the Nlrp3 inflamma- some confers protection against colorectal tumor formation.
J Immunol 2010;185:4912-4920.
97. Osaki T, Péron JM, Cai Q, et al. IFN-gamma-inducing fac- tor/IL-18 administration mediates IFN-gamma- and IL-12-in- dependent antitumor effects. J Immunol 1998;160:1742- 1749.
98. Osaki T, Hashimoto W, Gambotto A, et al. Potent antitumor ef- fects mediated by local expression of the mature form of the interferon-gamma inducing factor, interleukin-18 (IL-18).
Gene Ther 1999;6:808-815.
99. Cao E, Zang X, Ramagopal UA, et al. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent li- gand-binding surface. Immunity 2007;26:311-321.
100. Coughlin CM, Salhany KE, Wysocka M, et al. Interleukin-12 and interleukin-18 synergistically induce murine tumor re- gression which involves inhibition of angiogenesis. J Clin Invest 1998;101:1441-1452.
101. Hegardt P, Widegren B, Li L, et al. Nitric oxide synthase in- hibitor and IL-18 enhance the anti-tumor immune response of rats carrying an intrahepatic colon carcinoma. Cancer Immunol Immunother 2001;50:491-501.
102. Salcedo R, Worschech A, Cardone M, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med 2010;207:1625- 1636.
103. Hu B, Elinav E, Huber S, et al. Inflammation-induced tumori- genesis in the colon is regulated by caspase-1 and NLRC4.
Proc Natl Acad Sci U S A 2010;107:21635-21640.
104. Zaki MH, Lamkanfi M, Kanneganti TD. The Nlrp3 inflam- masome: contributions to intestinal homeostasis. Trends Immunol 2011;32:171-179.
105. Srivastava S, Salim N, Robertson MJ. Interleukin-18: biology and role in the immunotherapy of cancer. Curr Med Chem 2010;17:3353-3357.
106. Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mecha- nisms. Immunol Rev 2008;226:41-56.
107. Wilke CM, Wei S, Wang L, Kryczek I, Kao J, Zou W. Dual bio- logical effects of the cytokines interleukin-10 and interfer- on-γ. Cancer Immunol Immunother 2011;60:1529-1541.
108. Nava P, Koch S, Laukoetter MG, et al. Interferon-gamma regu- lates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity 2010;32:392- 402.
109. Günzel D, Florian P, Richter JF, et al. Restitution of single-cell defects in the mouse colon epithelium differs from that of cul- tured cells. Am J Physiol Regul Integr Comp Physiol 2006;
290:R1496-R1507.
110. Tong Q, Vassilieva EV, Ivanov AI, et al. Interferon-gamma in- hibits T84 epithelial cell migration by redirecting transcytosis of beta1 integrin from the migrating leading edge. J Immunol 2005;175:4030-4038.
111. Utech M, Ivanov AI, Samarin SN, et al. Mechanism of IFN-gam- ma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane.
Mol Biol Cell 2005;16:5040-5052.
112. Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-al- pha synergize to induce intestinal epithelial barrier dysfunc- tion by up-regulating myosin light chain kinase expression.
Am J Pathol 2005;166:409-419.