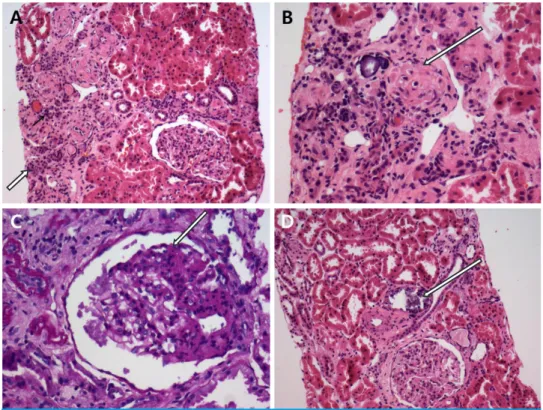
A Case of Secondary FSGS due to Chronic Chloride Diarrhea
5
0
0
전체 글
(2)
(3)
(4)
(5)
수치
관련 문서