Author contributions: K.S.H. and M.G.L. conceptualized the review study.
K.S.H. and M.G.L. contributed to writing and editing of the manuscript.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Copyright © Korean J Physiol Pharmacol, pISSN 1226-4512, eISSN 2093-3827
INTRODUCTION
The vascular endothelium is defined as a single layer of endo- thelial cells (ECs) that line the lumen of blood vessels and are me- chanically and metabolically dynamic organs. The endothelium, which consists of 1 – 6 × 10
13individual ECs, is the largest organ in the body and exceeds 1,000 m
2of estimated surface area [1-3].
This important organ is involved in a variety of physiological and pathological functions including blood supply, nutrient delivery,
immune cell adhesion, vasopermeability, angiogenesis, thrombo- genesis, and vascular tone [4-7].
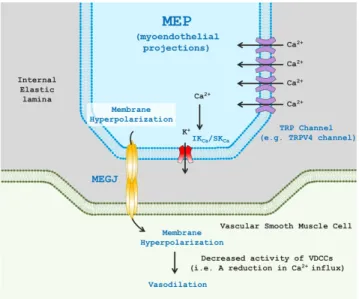
Endothelium-dependent vasodilation is largely determined by alterations in endothelial intracellular Ca
2+concentrations in response to mechanical stimuli (e.g., shear stress, membrane stretch) or endogenous agonists (e.g., bradykinin, ATP, or reactive oxygen species [ROS]). Increased intracellular Ca
2+levels produc- es nitric oxide (NO) and prostacyclin (PGI
2) that are traditionally considered as endothelium-derived relaxing factors (EDRFs) [8,9].
Review Article
Endothelial Ca 2+ signaling-dependent vasodilation through transient receptor potential channels
Kwang-Seok Hong 1 and Man-Gyoon Lee 2, *
1