개요
석유에너지 고갈, CO2에 의한 지구 온난화 문제 등 이 전 지구적 이슈가 되면서 화석연료의 대체 수단으 로 수소에너지와 이를 사용하는 연료전지 중요성이 강조 되고 있다. 수소 에너지 및 연료전지 보급 확대 를 위해서는 저가의 연료전지 소재(촉매, 전해질)를 개발하는 것이 필요하지만 기존의 개발 방법으로는 시간 및 비용의 제약 때문에 이를 달성하기가 쉽지 않 다. 따라서 연료 전지 소재 개발 비용 및 시간을 획기 적으로 줄일 수 있는 혁명적인 소재 개발 방법이 필요 하게 된다. 양자 역학의 기본 식인 슈뢰딩거 방정식을 최신의 First-principles density functional theory (DFT)에 의해서 풀게 되면 소재의 전자 구조 및 특 성을 비교적 정확하게 예측할 수 있게 된다. 따라서 연료전지 핵심 구성 요소인 촉매, 전해질의 기본 물성 에 대한 데이터베이스를 비용과 시간의 제약 없이 쉽 게 구축 할 수 있게 되고 향후 성능이 획기적으로 개 선된 연료전지 소재 개발을 가능하게 할 수 있다.
본 계산과학 특별기획에서는 DFT를 사용하여 연 료전지용 전이 금속 합금 촉매 설계에서 촉매 활성을 결정할 수 있는 다양한 합금 효과를 설명하고 산소환 원반응, 개미산 분해반응, CO2 환원 반응에서 이러한 합금 효과가 촉매 설계에 어떻게 적용될 수 있는지를 다룰 것이다.
나노 금속 합금 효과
최근의 연구결과[1-12]에 의하면 다성분 금속 촉매 의 반응성은 표면 원자의 분포[ensemble(혹은 geometric) 효과], 금속-금속 상호 작용에 의한 전자 구조의 변화 [ligand(혹은 electronic) 효과], 격자 상 수 불일치에 의한 촉매 활성 변화[strain effect]등 의 해서 결정될 수 있다고 알려져 왔다. 또한 반응성은 나노 촉매에서 표면 원자의 낮은 배위수(atom coordination)의 존재 여부, 표면에 노출되는 facet (111, 100, 110등) 종류, 및 나노 입자 형태 (icosahedron, octahedron) 등에 의해서 수정될 수 있 다고 보고되어 왔다(그림 1 참조). DFT 계산은 직접 그림 1. 촉매 활성을 지배할 수 있는 나노 금속 합금 효과
DFT(Density functional theory)를 통한 연료전지용 나노 금속 합금 촉매작용
(Nanometal alloy catalysis)의 이해
이상헌, 조진원, 함형철*
KIST 연료전지연구센터
분석이 가능하여 이러한 복잡한 합금 효과를 이해하 기 위한 강력하고 유연한 수단으로 사용될 수 있다.
AuPd 표면 합금에서 직접 H2O2 합성 반응: Pd monomer 중요성
Bimetallic Au-Pd 촉매의 직접 H2O2 반응성 (O2+H2 → H2O2)이 순수한 Au 혹은 Pd에 비해서 증진된다고 보고되어 왔다. 그러나 직접 분석의 어려 움 때문에 이에 대한 이해는 매우 부족한 실정이다.
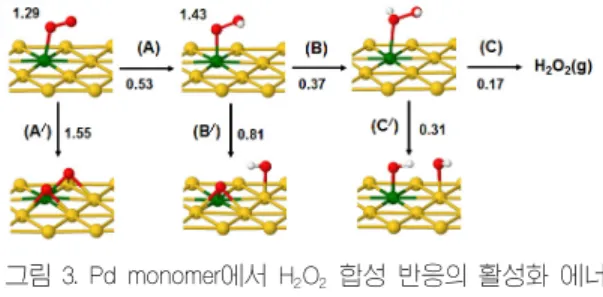
본 장에서는 AuPd(111) 촉매 표면에서 Pd 원자 분 포에 따른 표면 반응성을 이해하기 위해서 다양한 Pd ensembles(Au에 둘러싸여 있는 Pd 원자, 그림 2 참 조)위에서 직접 H2O2 합성 반응성(선택도)을 조사하 였다.
그림 2에 조사된 반응 단계를 나타내었다.(1) O2
hydrogenation [(A)] versus O + O scission [(A)/]; (2) OOH hydrogenation [(B)] versus O + OH scission [(B)/]; and H2O2desorption [(C)]
versus OH + OH scission [(C)/]. 표 1 에 hydrogenation 및 decomposition 반응에 대한 계산된 반응에너지(△E) 및 활성화 에너지(Ea)를 요약하였 다. 표에 보듯이 직접 H2O2 합성 반응성이 표면 Au 및 Pd 원자 배열에 크게 의존한다는 것을 예측하였고 특히 Pd(111) substrate에 지지된 주위의 Au로 완전 히 둘러싸여 있는 하나의 Pd 원자인 Pd monomer가 다른 크기의 ensembles(dimer, trimer 등)보다 H2O2
로의 선택도를 크게 증가 시킬 수 있다는 것을 알 수
있었다(그림 3 참조)[13,14].
이러한 DFT 예측(직접 H2O2 합성 반응성에서 Pd monomr의 중요성)은 Schiffrin 및 Han 등에 의해서 최근 실험으로 확인되었다[15,16].
이성분 Pd/Ag 촉매에서 개미산분해를 통한 수소 생산:Ligand effect의 중요성
Ag-Pd Core-Shell 촉매가 개미산으로부터 수소 발생에 순수한 Pd에 비해서 높은 활성을 나타낸다고 최근 보고가 되어왔다[17]. 그러나, Ag-Pd Core- Shell촉매의 활성 증가 메커니즘에 대해선 알려진 바 가 없다. 본 장에서는 DFT 계산을 통해 Ag-Pd Core-Shell 촉매에서 수소 생산 속도 및 선택도를 결 정하는 인자에 대해서 살펴 보았다.
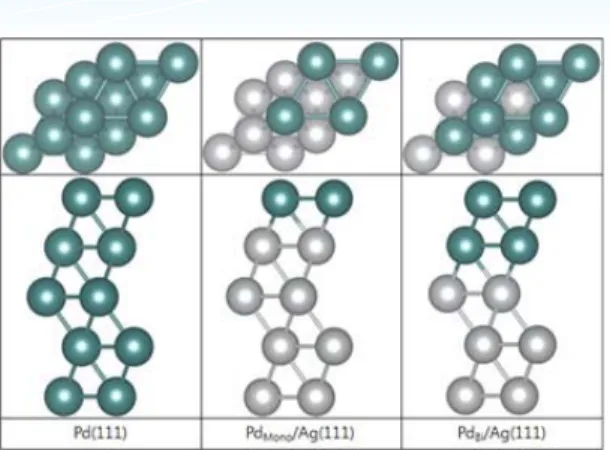
그림 4와 같이 각각의 bulk 모델을 FCC(111)으로 그림 2. AuPd(111) 표면 구조: Pd monomer (indicated as M),
dimer (D), trimer (T), and pure (P)
그림 3. Pd monomer에서 H2O2 합성 반응의 활성화 에너 지 및 중간 생선물의 분자 구조
표 1. 계산된 반응(△E) 및 활성화 에너지(괄호, Ea). 단 위는 eV임.
M D T P
(A)
O
2+H → OOH -0.73
(0.53) -0.25 (0.84) 0.00
(0.85) 0.18 (0.89) (A)/
O
2→ O+O 0.25 (1.55)
-0.23 (1.54)
-0.80 (1.05)
-2.08 (0.51) (B)
OOH+H → H
2O
2-0.86
(0.37) -0.51
(0.81) -0.20 (0.72) 0.09
(-)
a (B)/OOH → O+OH -0.39 (0.81)
-0.92 (0.54)
-1.29 (0.22)
-2.13 (0.31) (C)
H2O2→ H2O2(g) 0.17
(0.17) 0.18
(0.18) 0.21
(0.21) 0.24 (0.24) (C)/
H2O2→ OH+OH -0.90
(0.31) -1.35
(0.11) -1.58
(0.08) -2.09 (0.01)
잘라 Pd-shell의 두께에 따라 개미산 분해반응을 조 사하였다. 일반적으로 Pd 표면위에서 HCOOH의 분 해반응은 그림 5와 같이 두가지 반응으로 나누어지는 데, 하나는 HCOO+H를 거쳐 최종적으로 CO2와 H로 진행하여 수소를 얻는 반응식이 있는 반면, 다른 반응 은 HCO+OH를 거쳐 Pd 촉매에 독성을 띄우는 CO 와 물로 진행하는 반응식이 있다.
Pd 촉매에 있어 가장 이상적인 조건은 수소를 얻는 반응식에 대한 활성화 에너지가 낮고 동시에 CO로 진행하는 반응의 활성화 에너지는 높아야 한다. 그림
5에서 Pd-shell이 가장 얇은 PdMono/Ag(111)가 개미 산으로 수소를 생산하는 활성화 에너지가 가장 낮게 계산된 것을 확인할 수 있다[18,19]. 즉, PdMono/Ag(111) 촉매에서 온도가 높아질수록 Arrhenius 식에 입각하 여 수소 생산 속도 상수가 다른 촉매보다 높다는 것을 의미한다. PdBi/Ag(111)촉매 또한 Pd 촉매보다 수소 생산 속도가 개선 되었지만 CO로 가는 반응식에서 활성화 에너지가 Pd 촉매 보다 낮기 때문에 선택도 (ΔEselectivity=ΔEindirect-ΔEdirect)면에서 감소 하였다.
PdMono/Ag(111)는 좋은 촉매의 조건 중 하나인 CO 활성화 에너지가 다른 촉매와 비교해 가장 낮은 것으 로 계산되어 촉매의 이상적인 조건을 다 갖추었다. 이 와 같이 Pd-shell의 두께가 바뀌면서 수소 생산성 및 선택도 또한 크게 바뀐걸 알 수 있었는데 그 이유를 다음에 보다 더 자세히 다루었다.
2장에서 설명한 다양한 합금 효과 중에서 Strain 및 ligand effect가 Pd/Ag core-shell 촉매의 활성을 결 정하는 중요한 역할을 한다. 첫 번째로 strain effect는 Pd원자와 Ag원자의 격자상수가 서로 달라 표면의 Pd가 substrate(혹은 bulk, core 부분)의 Ag의 격자 상수를 갖게 되면서 생기는 현상이고 다른 하나는 ligand effect는 표면의 Pd원자와 표면아래의 Ag원 자가 서로 상호작용을 하여 Pd의 전자 구조가 바뀌어 표면 활성이 바뀌는 현상이다. 보통 두 합금 효과는 동시에 발생하므로 실험으로 각각의 효과가 촉매에 그림 4. 왼쪽부터 Pd, Pd-shell이 monolayer인 PdMono/Ag(111)
및 bilayer 인 PdBi/Ag(111)의 표면 구조
그림 5. Direct Pathway는 수소 생산 반응이고 Indirect Pathway는 CO 생산 반응이다. 그래프는 수소 생산을 rate 을 나타내는 Productivity와 Selectivity를 나타낸다.
그림 6. Strain effect와 Ligand Effect
미치는 영향을 조사하기는 불가능하나 계산과학의 장 점을 살려 각 각의 효과를 서로 분리하여 영향력을 계 산 할 수 있다. 단일원자로 이루어진 촉매에서는 격자 상수가 클수록 흡착에너지가 증가한다고 알려져 있는 데(tensile strain effect), Pd 보다 격자상수가 큰 Pd/Ag에서는 오히려 흡착에너지가 줄어든 걸 발견 할 수 있었다. 이러한 결과를 strain effect 관점에서 계산하였다. 예를 들면, strain effect의 계산은, PdMono/Ag(111)모델에서 Ag를 Pd로 치환하여 그 림 6과 같이 겉보기엔 Pd와 같이 보이지만 사실 Pd 가 Ag의 격자상수를 갖게 되는 모델을 만들어
(PdStrained라고 칭함) 개미산의 분해반응의 중요 인자
로 알려진 HCOO를 흡착하여 binding energy를 구하 면, 비로소 다음과 같은 식을 통해 strain effect를 계 산할 수 있다. ΔEHCOOstraineffect=ΔEHCOO,Pdmono/Ag-Δ EHCOO,Pdstrained. 계산 결과 Pdstrain의 HCOO binding 에 너지는 Pd HCOO binding energy 보다 증가한 걸 확인하였고 즉, strain effect는 활성이 감소된 Pd/Ag 표면의 현상을 설명하기 충분하지 않은 걸 알 수 있었 다. 그렇다면, ligand effect 때문에 표면 활성이 줄었 다고 할 수 밖에 없는데 이것을 확인하기 위해 다음과 같은 식을 통해 ligand effect를 계산하였다. Δ Ead,ligand_effect=ΔEad,Pdmono/Ag-ΔEad,Pd-ΔEad,Pdstained. 계산 결과 PdMono/Ag(111)의 ligand effect는 strain effect 보다 훨씬 더 크게 작용하는 걸 알 수 있었다. 즉, Pd- Ag의 ligand effect가 binding energy를 증가시키는 strain effect보다 강하게 작용하여 HCOO binding energy를 감소시킨 것을 확인하였다. 그러면 왜 ligand effect가 PdMono/Ag(111)에서 두드러지게 나 타난 이유에 대해서도 Charge Transfer(Bader charge anlaysis)라는 계산을 통하여 조사하였다(전 자의 이동방향과 그 양을 계산). 계산결과, ligand effect의 크기와 전자가 표면으로 이동한 양이 비례하 였다. PdMono/Ag(111)에서 Pd표면은 다른 두 모델에 비해 Ag로부터 가장 많은 전자를 얻은 걸 확인하였 고 (ligand effect 또한 다른 모델에 비해 압도적으로
크다), 표면으로 전자가 많이 이동한 순으로 수소 생 산 활성화 에너지가 낮다는 것을 알 수 있었다. 즉, 전 자가 표면으로 이동하여 표면의 전자구조를 바꾸어 binding energy를 줄였다는 것뿐만 아니라 Pd-shell 이 가장 얇을 때 수소 생산 속도와 선택도가 가장 좋 다는 것을 확인할 수 있었다.
전기화학적 CO2환원 기술
탄소를 기반으로 한 화합물들 중에서 일반적인 환 경에서 가장 안정한 물질인 CO2는 전기화학적인 환원 법을 통하여 유용한 화합물들로 전환될 수 있다. 에너 지 변환 관점에서 이 기술의 파급 효과를 극대화하기 위해서는, 적은 양의 전기에너지를 사용하여 부산물 의 생성을 억제하면서 원하는 생성물을 최대한 얻어 내는 기술이 필요하다. 이는 CO2 환원반응이 일어나 는 금속 표면의 구조를 효과적으로 제어하면서 이루 어질 수 있다. 일반적으로 전기화학적 CO2 환원반응 을 통한 탄화수소 합성에 가장 효과적인 금속 촉매로 알려진 구리(Cu)의 경우, 아래와 같은 환원반응을 통 하여 CH4를 합성하는데 열역학적으로 +0.17 V(vs.
RHE, 18.5˚C)의 전기에너지가 필요하다.
CO2+8(H++e-)→ CH4+2H2O (1)
하지만 실제 실험에서는 -1.0 V의 높은 전기에너지 가 필요 하다[20]. 이러한 차이에 대한 의문을 해소하 고 전기화학적 CO2환원반응에 대한 상세메커니즘을 제공하기 위한 이론적인 방법으로 CHE 모델 (Computational Hydrogen Electrode Model)이 개 발되었다[21]. 이 모델에서는 전체 CO2환원반응 경로 를 각각의 단위 반응 단계로 세분화하고(그림 7 참조) 이들 단위반응에 대한 자유에너지(Gibbs Free Energy) 변화를 계산한다. 그리고 모든 단위반응에 대해서 자유에너지들이 줄어드는 방향으로 변할 경우 비로소 전체 CO2환원반응이 진행될 수 있다고 가정 한다. 각 단계별 자유에너지[ΔGn,n=(H++e-)의 수]
는 흡착물의 흡착에너지를 이용해서 얻어지고 적용된 전기에너지(U)는 ΔGn(U)=ΔGn(U=0) + neU (e=단일 전자 전하)의 형태로 ΔGn에 포함된다. 흡 착에너지는 일반적으로 DFT 계산을 통하여 얻어진 다.
CHE 모델을 CO2 환원용 Pd/Au 합금 촉매 개발 에 적용해보았다. Pd/Au 촉매의 경우 촉매 제조 조 건을 조절함으로써 촉매 표면의 Pd/Au 조성의 조절 이 가능하다[6]. CHE 모델은 촉매 표면 조성의 변화 가 반응 메커니즘에 어떤 영향을 미치는지에 대한 이 론적인 근거를 제공해 줄 수 있다. 그림 8(a)는 다양 한 금속 표면에서 계산된 CO2 환원반응 경로에 따른 자유에너지 변화(전기에너지를 가하지 않은 경우)를 요약해서 보여준다. 이들의 경우 열역학적으로 CO2→ CH4 전체 반응은 자발적이지만 각각의 단위반응들은 자발적인 반응과 비자발적인 반응이 섞여 있다는 것 을 보여준다. 하지만 그림 8(b) Cu 촉매의 경우에서 보이는 봐와 같이 일정 크기 이상의 전기에너지(U = -0.91 V)가 가해지면 모든 단위 반응들이 자발적으로 전환되면서 비로소 CO2 → CH4 전체 반응이 진행될 수 있다는 것을 보여준다. 그리고 CO2가 한 개의 (H++e-)를 받아서 COOH 그룹 형태로 Cu 표면에 흡착하는 단위반응이 전체 반응 속도를 결정하는 단 계(Rate Determining Step)라는 것을 알 수 있다. 그 리고 Pd/Au 합금 촉매들의 경우, Cu와 순수 Au 촉
매에 비해서 CO2 → COOH* 반응의 반응에 필요한 에너지가 크게 줄어드는 대신에 CO* → CHO* 반응 에 필요한 에너지는 크게 증가한다. 이러한 반응에너 지의 변화 정도는 표면에서 Pd 조성이 증가할수록 더 욱 심해져서 CO* → CHO* 반응이 속도 결정 단계가 된다. 이 계산 결과는 Pd 원자들과 주변의 Au 원자들 의 상호작용에 의해서 Pd 활성점들의 반응성이 제어 되고, 궁극적으로 CO2환원반응 전체 반응 속도와 선 택성에 영향을 미칠 수 있다는 것을 보여준다[22]. 이 와 같은 CHE 모델을 이용하면 CO2 환원 반응에 대 그림 7. 환원 단계별 CO2환원반응 경로
그림 8. CHE 모델에 의하여 계산된 CO2환원반응 경로에 따른 자유에너지 변화. (a) 다양한 금속 표면에서 전기에 너지를 가하지 않을 경우(U=0), (b) Cu 표면에 전기에너 지 (U=-0.91 V)를 가할 경우. 모든 단위반응의 자유에너지 변화가 줄어드는 방향으로 나타난다. 즉, Cu 표면에서 CO2환원반응이 일어나기 위해서는 -0.91 V의 전기에너지 가 필요하다.
한 다양한 금속 촉매들의 반응성과 선택성에 대한 탐 색이 가능하며, 실험 연구와 적절히 결합될 경우 저비 용·고효율의 신(新)촉매 개발에 드는 시간과 비용을 획기적으로 줄일 수 있다.
맺는말
양자 역학에 기반을 둔 DFT 계산은 연료전지 소재 의 물리/화학적 특성을 예측하고 소재 구조와 실제 연 료전지 성능과의 관계 모델을 개발하는 것이다. 따라 서 최적의 성능을 갖는 소재 선정을 위한 가이드라인 을 제시 할 수 있게 된다. 또한, DFT 계산을 통한 소 재 연구/개발은 정보의 투명성 및 통합성을 높일 수 있어 소재 개발 기간을 크게 줄일 수 있어 향후 성능 이 개선된 연료전지 소재 개발을 가능하게 할 수 있다.
참고문헌
1. H. C. Ham, D. Manogaran, K. H. Lee, K. Kwon, S. - A. Jin, D. J.You, C. Pak and G. S. Hwang, J. Chem.
Phys-Communications., 113399, 201104(2013)
2. M. Pan, A. J. Brush, D. Pozun, H. C. Ham, W.-Y. Yu, G. Henkelman, G. S. Hwang and C. B. Mullins, Chem.
Soc. Rev., 4422, 5002(2013)
3. M. Pan, H. C. Ham, W- Y. Yu, G. S. Hwang and C.
B. Mullins, J. Am. Chem. Soc., 113355, 436(2013) 4. B. Patrick, H. C. Ham, Y. Shao-Horn, L. F. Allard, G.
S. Hwang and P. J. Ferreira, Chem. Mater., 2255((44)), 530(2013)
5. K.-S. Lee, H.-Y. Park, H. C. Ham, S. J. Yoo, H. J.
Kim, E. Cho, A. Manthiram and J. H. Jang, J. Phys.
Chem. C., 111177((1188)), 9164(2013)
6. S.-Y. Lee, Y.-H. Park, J. W. Cho, H. C. Ham, J. H.
Jang and S. J. Yoo, ACS Catal., 44, 2402(2014) 7. Martin, D. H. Lim, H. V. P. Nguyen, S. K. Kim, S. P.
Yoon, J. Han, S. W. Nam, C. W. Yoon and H. C.
Ham, Int. J. Hydrogen Energy., 3399((2233)), 12251(2014)
8. D. Y. Chung, S.-K. Park, Y.-H. Chung, S.-H. Yu, D.- H. Lim, N. Jung, H. C. Ham, H.-Y. Park, Y. Piao, S.
J. Yoo and Y.-E. Sung,Nanoscale., 66, 2131(2014).
9. J. A. Stephenson, H. C. Ham and G. S. Hwang, J.
Phys. Chem. C., 111144, 21516(2010)
10. H. C. Ham, J. A. Stephenson, G. S. Hwang, J. Han, S. W. Nam and T. H. Lim, Catalysis Today., 116655, 138(2011)
11. H. C. Ham, J. A. Stephenson, G. S. Hwang, J. Han, S. W. Nam and T. H. Lim, J. Phys. Chem. Lett., 33, 566(2012)
12. H. C. Ham, G. S. Hwang, J. Han, S. W.Nam and T.
H. Lim, Surf. Sci., Submitted for publication(2014) 13. H. C. Ham, G. S. Hwang, J. Han, S. W. Nam and T.
H. Lim, J. Phys. Chem. C-Letter., 111133, 12943(2009) 14. H. C. Ham, G. S. Hwang, J. Han, S. W. Nam and T.
H. Lim, J. Phys. Chem. C., 111144, 14922(2010) 15. J. S. Jirkovsky´, I. Panas, E. Ahlberg, M. Halasa, S.
Romani and D. J. Schiffrin, J. Am. Chem. Soc., 113333((4488)), 19432(2011)
16. L. Ouyang, G.-j. Da, P.-f. Tian, T.-y. Chen, G.-d.
Liang, J. Xu and Y.-F. Han, J. Catal., 331111, 129(2014) 17. K. Tedsree, T. Li, S. Jones, C. W. Chan, K. Yu, M.
Bagot, E. A. Marquis, G. D. Smith and S. C. Tsang, Nat. Nanotechnol., 66(5), 302(2011)
18. J. Cho, S. H. Lee, J. Han, S. P. Yoon, S. W. Nam, S.
H. Choi, K. Y. Lee and H. C. Ham, J. Phys. Chem.
C., 111188, 22254(2014)
19. J. Cho, S. H. Lee, J. Han, S. W. Nam, K. Y. Lee and H. C. Ham, Curr. Appl. Phys., submitted for publication(2014)
20. Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc.
Fraday Trans., 11, 85, d2309(1989)
21. A. A. Peterson, F. Abild-Pedersen, F. Studt, J.
Rossmeisl and J. K. Nø rskov, Energy &
Environmental. Sci., 33, 1311(2010)
22. S. H. Lee, J. H. Jang, J. Han, S. W. Nam and H. C.
Ham, In preparation(2014)