http://dx.doi.org/10.20307/nps.2015.21.4.231
231
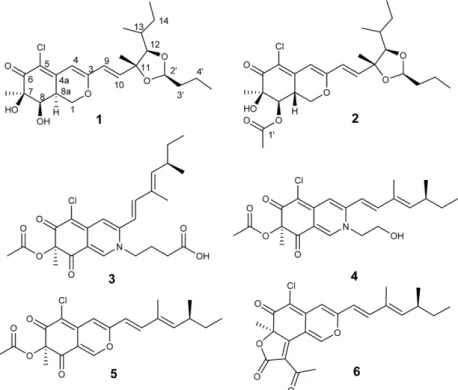
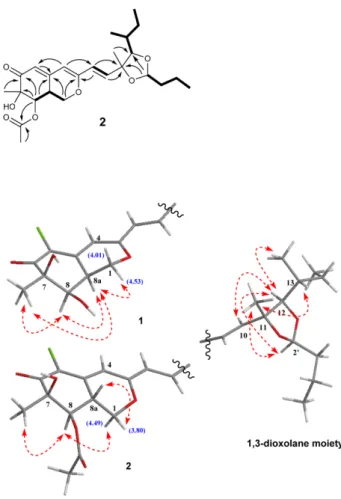
Penidioxolanes A and B, 1,3-Dioxolane Containing Azaphilone Derivatives from Marine-derived Penicillium sp. KCB12C078
Seung Min Kim 1 , Sangkeun Son 1,2 , Jong Won Kim 1 , Eun Soo Jeon 1 , Sung-Kyun Ko 1 , In-Ja Ryoo 1 , Kee-Sun Shin 5 , Hiroshi Hirota 3 , Shunji Takahashi 3,4 , Hiroyuki Osada 4 , Jae-Hyuk Jang 1,2, *, and Jong Seog Ahn 1,2, *
1
Chemical Biology Research Center, Korea Research Institute of Bioscience and Biotechnology, Chungbuk 363-883, Korea
2
Department of Biomolecular Science, University of Science and Technology, Daejeon 305-333, Korea
3
RIKEN-KRIBB Joint Research Unit, Global Research Cluster, RIKEN, Saitama 351-0198, Japan
4
Chemical Biology Research Group, RIKEN CSRS, Saitama 351-0198, Japan
5