EBV-Associated Lymphoproliferative Disorders
Young Hyeh Ko
1,2Department of Pathology,
1Korea University Guro Hospital and
2Hanyang University Hospital, Seoul, Korea
Epstein–Barr virus (EBV) is associated with a wide range of human lymphoprolifer- ative disorders (LPD) of B, T, and natural killer (NK)-cell lineage. In children, abnor- mal immune response to primary EBV infection can cause peculiar forms of T/NK- cell LPD of childhood, such as the systemic form of chronic active EBV infection, hydroa vacciniforme-like LPD, severe mosquito bite allergy and systemic T cell lym- phoma of childhood. In adults, dysregulation of the immune response to EBV in- fection, immunosenescence caused by aging, chronic inflammation in a closed space, and iatrogenic immune suppression can lead to EBV-positive LPD of diverse types involving B, T, and NK cells with unique clinical and pathological presentations. This review describes the clinical, pathological, and genetic findings of EBV-positive LPD listed in the revised 2016 WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue.
pISSN 2233-5250 / eISSN 2233-4580 https://doi.org/10.15264/cpho.2021.28.1.14
Clin Pediatr Hematol Oncol 2021;28:14∼27Received on October 5, 2020 Revised on December 11, 2020 Accepted on December 31, 2020
Corresponding Author:
Young Hyeh Ko
Department of Pathology, Korea University Guro Hospital, 148 Gurodong-ro, Guro-gu, Seoul 08308, Korea
Tel: +82-2-2626-1482 Fax: +82-2-2626-1486 E-mail: [email protected] ORCID ID: orcid.org/0000-0002-4383-0579 Key Words: Epstein-Barr virus, Lymphoma, Lymphoproliferative disorder
Copyright ⓒ 2021 Korean Society of Pediatric Hematology-Oncology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/
Introduction
Epstein–Barr virus (EBV) was the first virus shown to cause cancer in humans and is associated with a wide range of human lymphoproliferative diseases (LPDs). EBV infects primarily B cells via the EBV receptor on the B-cell surface, although EBV infrequently also infects T or NK cells. Depending on host immunity and viral fac- tors, viral persistence in host cells can induce lympho- proliferation with a diverse clinical spectrum ranging from simple reactive hyperplasia to aggressive lympho- ma/leukemia with unique clinical and pathological presentations. In children, EBV infection in T or NK cells causes peculiar forms of T/NK-cell LPDs, such as the systemic form of chronic active EBV infection (CAEBV), hydroa vacciniforme (HV)-like LPD, severe mosquito bite
allergy and systemic T cell lymphoma of childhood as listed in the recent WHO classification [1]. This review describes the clinical, pathological, and genetic findings related to EBV-positive LPD, in which EBV positivity is an essential component for diagnosis. The Burkitt and Hodgkin lymphomas, in which EBV is found in a subset of tumors, are not included (Table 1).
EBV-Positive Lymphoproliferative Disorders in Childhood
1) Chronic active EBV infection, systemic form
Primary EBV infection usually regresses spontaneously
but often results in infectious mononucleosis, which is
usually resolved within 2 weeks after onset but can per-
sist for a month or, in rare cases, even longer. Rarely,
patients who have primary EBV infection or, less com-
Table 1. Classification of EBV-positive lymphoproliferative disorders
Diagnosis Main age group Pathogenesis EBV infected cell Clinical course Chronic active EBV
infection, B, systemic Children and
young adults Genetic defect in host immune
response to EBV B-cells Variable from indolent to aggressive
Chronic active EBV
infection, T & NK, systemic Children and
young adults Genetic defect in host immune
response to EBV T or NK cells Variable from indolent to aggressive
Severe mosquito-bite allergy Children and
young adults Genetic defect in host immune
response to EBV Mainly NK cells Variable from indolent to aggressive
Hydroa-vacciniforme-like
LPD Children and
young adults Genetic defect in host immune
response to EBV T cells Variable from indolent to aggressive
Systemic EBV-positive T cell
lymphoma of childhood Children Genetic defect in host immune
response to EBV T cells Fulminant
EBV-positive DLBCL, NOS Elderly and
young adults Age-related immune deficiency
in the elderly B-cells Depends on age;
aggressive in the elderly EBV-positive mucocutaneous
ulcer Elderly Iatrogenic or age-related
immune deficiency B-cells Mostly benign DLBCL associated with
chronic inflammation Adults Local immunodeficiency in a
closed space B-cells Aggressive
Fibrin-associated DLBCL Adults Local immunodeficiency in a
closed space B-cells Mostly benign
Lymphomatoid
granulomatosis Adults Underlying immunodeficiency B-cells Variable depending on grade
Extranodal NK/T cell
lymphoma, nasal-type Adults Oncogenic potential of EBV NK or T cells Aggressive Aggressive NK cell leukemia Adults Oncogenic potential of EBV NK cells Fulminant PTCL, NOS, EBV-positive Adults Immune deficiency
Oncogenic potential of EBV T cells Aggressive
LPD, lymphoproliferative disorders; DLBCL, diffuse large B cell lymphoma; PTCL, peripheral T cell lymphoma; NOS, not otherwise specified.
monly, EBV reactivation, develop CAEBV, a form of EBV-positive LPD. CAEBV, systemic form is characterized by fever, persistent hepatitis, hepatosplenomegaly, and lymphadenopathy, which show varying degrees of se- verity depending on the host immune response and EBV viral load [2]. The diagnostic criteria for CAEBV include chronic illness lasting at least 3 months, increased EBV DNA in either the tissue or blood mononuclear cells (>10
2.5copies/ μ g), and lack of evidence of a known un- derlying immunodeficiency according to the recent WHO classification [1].
CAEBV is a rare disease and has strong racial pre- disposition. It is reported more often in Asian countries, such as Japan, Korea, and China, and in Latin America, including South and Central America, and Mexico, than in Europe and North America (Canada and the United States). The disease develops in children and adolescents
but can also develop in young adults and elderly patients, although with a lower frequency [3].
CAEBV in Asia and Latin America affects T or NK cells, and B-cell CAEBV accounts only for 3% of CAEBV in Asia [4]. CAEBV is much rarer in Western populations than in Eastern populations. In the largest series of CAEBV re- ported in the United States, nine of 10 patients, exclud- ing Hispanics and Asians, had CAEBV of the B-cell type, while only one patient had T-cell type CAEBV [5]. Most patients with CAEBV T/NK-cell-type LPD have no con- sistent immunological abnormality, although B-cell CAEBV most often involves a progressive loss of B cells and hy- pogammaglobulinemia [5]. Reduced NK activity [6] and impaired EBV-specific CTL activity [7] have been reported in CAEBV-T and NK.
The higher prevalence of CAEBV-T/NK in Asians or
indigenous Central and South Americans suggests that
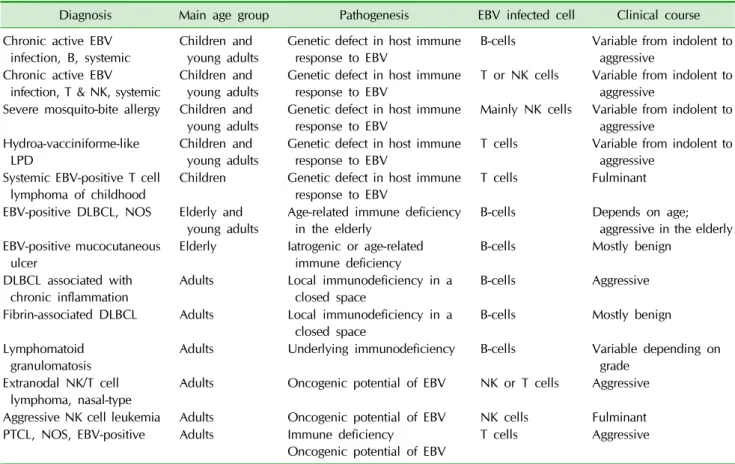
Fig. 1. (A) Chronic active EBV infection, T/NK cell. Liver biopsy shows minimal histologic change. (B) EBER in situ hybridization highlights EBV infected lymphocytes in hepatic sinusoids.
genetic factors that control the immune response to EBV infection underlie the susceptibility to the development of EBV-positive T/NK-cell LPD in young children and adolescents. Recent comprehensive genetic analysis us- ing whole-exome sequencing has shown that the EBV ge- nome in patients with CAEBV T/NK-cell or EBV-asso- ciated lymphoma harbor frequent intragenic deletions, which suggests a unique role of these mutations in the neoplastic proliferation of EBV-infected cells. In addi- tion, somatic driver mutations are frequently found in DDX3X, KMT2D, BCOR, BCORL1, TET2, and KDM6A. These mutations were also observed in extranodal NK/T cell lymphoma, a prototype of EBV-positive T/NK cell lym- phoma, suggesting that a common molecular mechanism acts in the process of clonal evolutions of EBV-infected T or NK cells [8].
Abnormal activation and replication of EBV together with the proliferation and clonal expansion of infected cells play a key role in the pathogenesis of CAEBV. The proliferating cells lack histological evidence of malig- nancy and may be polyclonal, oligoclonal, or monoclonal according to the stage of transformation [1,3]. Clonality does not necessarily indicate a worse prognosis.
The clinical manifestations of CAEBV vary. In addition to fever, chronic hepatitis, and lymphadenopathy, pa- tients frequently show NK lymphocytosis and, less com- monly, bowel perforation, coronary artery aneurysms, immunoglobulin A nephropathy, choreic movement, brain infarction, mosquito bite hypersensitivity, or HV-like erup- tion [3,4]. CAEBV is diagnosed based on the clinical and laboratory findings. Pathological changes are nonspecific except for identification of EBV- infected T or NK cells in the biopsy tissue (Fig. 1) [1]. CAEBV has significant overlap in clinical manifestation with EBV-HLH and sys- temic EBV-positive T cell lymphoma. Patients with CAEBV are often complicated by EBV-associated hemophgocytic syndrome and may progress to systemic T cell lymphoma during the course of disease.
Patients with CAEBV die of infection, hemophagocytic syndrome, or progressive lymphoproliferation. The only proven effective treatment for this disease is hema- topoietic stem cell transplantation (HSCT). In a Japanese
study of patients with T or NK cell CAEBV, the 3-year overall survival rates in patients treated with chemo- therapy only, chemotherapy followed by allogeneic HSCT, or allo-HSCT only were 0%, 65%, and 82%, respectively [9]. Factors indicating a poor prognosis include late onset of disease, thrombocytopenia, and T cell CAEBV [4]. The prognosis of patients with B-cell CAEBV is similar to that of patients with T/NK-cell CAEBV, and only two of five patients with B-cell CAEBV were alive after HSCT [5].
2) Severe mosquito bite allergy
Mosquito bite reaction is common in the general pop- ulation and usually raises no specific medical problems.
Severe mosquito bite allergy, also known as mosquito bite hypersensitivity, is defined as an EBV-positive NK-cell LPD with peculiar cutaneous manifestations characterized by intense local skin symptoms, including erythema, bullae, ulcers, and skin necrosis following a mosquito bite. This condition may also have other symp- toms such as fever, malaise, hematuria, NK lymphocy- tosis, and abdominal cramps [1,10,11]. Mosquito bite al- lergy is not a simple allergic reaction but a cutaneous manifestation of underlying EBV-positive NK-cell pro- liferative disease [12]. EBV-infected NK cells are mono- clonal or oligoclonal according to EBV terminal repeat analysis, which suggests that patients with severe mos- quito bite allergy have clonal NK cell proliferation [13].
The geographic distribution of mosquito bite allergy is
similar to that of other EBV-positive T/NK-cell lympho-
proliferative diseases. Most patients are in the first two
decades of life, with a median age of 6.7 years (range
0-18 years) [13]. Patients usually experience repeated
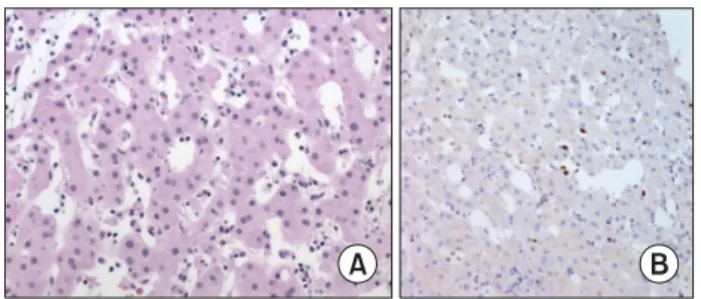
Fig. 2. (A, B) Severe mosquito bite allergy. Skin biopsy shows epi- dermal necrosis with perivascular cellular infiltration in the dermis.
(C) EBER in situ hybridization re- veals EBV infected lymphocytes.
episodes of severe symptoms induced by mosquito bites.
Skin lesions of severe mosquito bite allergy can be trig- gered by injection of mosquito saliva, which results in prominent infiltration of CD4+ T cells. In vitro studies have suggested that CD4+ T cells stimulated by mosquito bites play a key role in the development of the skin le- sions and that NK cell oncogenesis occurs via re- activation of EBV in latently infected NK cells and EBV oncogene expression [14,15]
Patients have a high serum IgE level and high EBV load in the peripheral blood. The skin at the mosquito bite site shows epidermal necrosis and ulceration (Fig. 2).
Predominant infiltration in the skin is CD4+ T cells with some CD8+ T cells and EBV-positive NK cells [1]. Al- though NK-cell lymphocytosis is common in the periph- eral blood, EBV-positive NK cells comprise a minor pop- ulation of the cells in the skin infiltrate [16].
The clinical course of mosquito bite allergy is variable.
Some patients have a prolonged and indolent disease course, which may be complicated by T/NK-cell CAEBV or HV-like eruptions. Half of patients die of hemophago- cytic syndrome or aggressive NK-cell leukemia (ANKL) [13,16,17].
3) Hydroa vacciniforme-like LPD
HV-like LPD is a peculiar cutaneous form of chronic EBV-positive LPD of polyclonal or monoclonal T cells and displays a broad spectrum of clinical aggressiveness and usually a long clinical course [1]. This disease has been reported under various names including angiocen- tric cutaneous T cell lymphoma of childhood [18], edem- atous scarring, vasculitic panniculitis [19], EBV-asso-
ciated lymphoproliferative lesion with HV-like eruption [20], classic HV and severe HV [21], or HV-like lymphoma [22], depending on severity of the cutaneous manifest- ations. The revised 2016 WHO classification proposed the term “HV-like LPD” to include the full spectrum of HV-like eruption because of the broad clinical spectrum of the disease and the lack of reliable morphological and molecular criteria for predicting its clinical behavior [1].
The geographic distribution of this disease is similar to that of other EBV-positive T/NK-cell LPD and is more common in children in Asia and Latin America. The me- dian patient age at diagnosis is 8 years [1]. HV-like LPD can develop in adults, but the frequency is low [23,24].
HV-like LPD is characterized by blistering photo- dermatosis in childhood that heals with vacciniform scarring. The severity of skin lesions varies according to season and UV ray exposure. The number of EBV-pos- itive lymphocytes increases in spring and summer, but few EBV-positive cells are present during periods of re- mission in autumn and winter [16]. This observation sug- gests that EBV-infected T or NK cells are recruited to the skin and activated following sun exposure. Local pro- duction of interferon-gamma and chemokines by cellular infiltrates results in inflammation and tissue damage [25]
which lead to epidermal reticular degeneration and spongiotic vesiculation (Fig. 3).
Histologically, the dermis contains perivascular and
periappendiceal lymphocytic infiltration. Most of the in-
filtrating cells are CD4+ or CD8+ T cells, and NK cells
are rare. Most cells express cytotoxic molecules such as
TIA-1 and granzyme B. The histological changes of se-
vere HV are similar to those of classic HV, but the dermal
Fig. 3. (A) Hydroa vacciniforme-like lymphoproliferative dis- order. Skin biopsy shows vesicular degeneration of the epi- dermis and lymphocytic infiltration in the dermis. (B) Numerous EBV infected lymphocytes present.
infiltrates tend to be more extensive and deeper, and can reach to the subcutaneous tissue [16].
As in other types of EBV-positive T/NK-cell LPD, HV-like LPD is uncommon in Caucasians. A recent series from the United States reported on the clinicopatho- logical findings of HV-like LPD in 10 Caucasian patients.
Whites with HV-like LPD were less likely to experience systemic disease and had a much better prognosis than nonwhites [26]. The higher prevalence of severe HV in persons of Asian descent may be ascribed to the influ- ence of the genetic background and may be linked to the HLA type or environmental factors and immunological tolerance because of early exposure to EBV infection [27].
HV-like LPD is classified into two types based on the clinical features. The classic type is a self-limited form characterized by the formation of vesicles on sun-ex- posed areas, and the lesions heal with vacciniform scar- ring after photoprotection. All patients show photo- sensitivity either as a positive reaction to UV provocation or symptoms induced by sun exposure. These patients have a high EBV viral load in the tissue and peripheral blood, but evidence of systemic involvement is usually lacking [21]. In most patients, this condition tends to pursue a benign course, although chronic recurrent erup- tion can result in severe scarring and disfiguration of the face. However, one-third of patients with classic HV-like LPD can develop cutaneous or systemic T-cell lymphoma or systemic CAEBV 3-19 years after the initial presen- tation. The severe type of HV-like LPD involves more ex-
tensive skin lesions that occur on both sun-exposed and -unexposed skin. This form is associated with systemic manifestations including fever, hepatomegaly, serologic abnormalities, and peripheral NK lymphocytosis. Half of cases of the severe form progress to EBV-associated T-cell malignancy. For patients with HV-like LPD with systemic symptoms, only HSCT has been reported to be curative [28].
4) Systemic EBV-positive T cell lymphoma of childhood Systemic T-cell lymphoma of childhood is a major cat- egory of EBV-positive T/NK cell LPD of childhood and is a life-threatening illness that occurs in children. This disease is characterized by the systemic clonal pro- liferation of T cells, and the characteristic clinical fea- tures including acute onset of fever, hemophagocytic syndrome, pancytopenia, and organomegaly. The clinical progression is rapid, and patients die of sepsis, intra- vascular coagulation, and multiorgan failure usually within days or weeks after the diagnosis [1,16,22].
Systemic T-cell lymphoma of childhood has been de- scribed under a variety of terms including fatal EBV-as- sociated hemophagocytic syndrome [29], fulminant EBV- positive T cell proliferative disorder of childhood [30], or fatal hemophagocytic lymphohistiocytosis (HLH) [31].
This condition usually occurs in children with primary EBV infection or during the course of systemic CAEBV infection. The infection of T cells by EBV activates T cells to secrete Th1 cytokines such as tumor necrosis fac- tor-alpha and IFN-gamma, which subsequently activate macrophages [32].
The distinction between EBV–HLH and systemic T-cell LPD is challenging in the diagnosis of EBV-positive T/
NK-cell LPD [33,34]. In the revised 2016 WHO classi- fication, EBV–HLH is included as part of the systemic form of EBV-positive T-cell LPD and is considered to be a self-limited nonneoplastic hyperimmune reaction.
These two categories share similar histological and im-
munophenotypic findings. Excluding systemic T-cell lym-
phoma arising in patients with CAEBV, most patients
with EBV-positive systemic T-cell lymphoma are initially
diagnosed with EBV-associated HLH [33]. Cells infiltrat-
Fig. 4. (A) Systemic T cell lymphoma of childhood. Bone marrow biopsy shows many hemophagocytic histiocytes. (B) Lymph node biopsy shows infiltration of EBV-positive atypical T-lymphocytes with necrosis. (C) CD3. (D) EBER in situ hybridization.
ing into the bone marrow and tissue lack obvious atypia both in EBV–HLH and initial biopsy of systemic T-cell lymphoma (Fig. 4A). The immunophenotype of the in- filtrating cells is CD8+ cytotoxic T cells in both con- ditions. Systemic T-cell lymphoma is a clonal disease by definition of the 2016 revised WHO classification. Like- wise, EBV-HLH can be clonal [35]. There are reports that patients initially diagnosed with EBV–HLH have died of systemic T-cell lymphoma after treatment with the HLH- 2004 regimen [33]. In these patients, the initial biopsy tissue lacked cytological atypia, although subsequent bi- opsy showed atypical EBV-positive T lymphocytes in- filtrated into the tissue along with necrosis (Fig. 4B-D).
These overlapping clinical and pathological findings sug- gest that EBV–HLH and systemic T-cell lymphoma of childhood reflect a biological continuum rather than dis- crete entities [33,34].
EBV-Positive Lymphoproliferative Disorders in Adulthood
1) EBV-positive diffuse large B-cell lymphoma, not otherwise specified
EBV-positive diffuse large B-cell lymphoma, not oth- erwise specified (DLBCL, NOS) is an EBV-positive clonal B cell proliferation which can be diagnosed after ex- clusion of specific type of EBV positive B cell pro- liferation such as lymphomatoid granulomatosis (LYG) and DLBCL associated with chronic inflammation. It was initially defined as an aggressive large B cell lymphoma
arising in patients older than 50 years [22], however, a substantial proportion of EBV-positive DLBCL can occur in young adults [36,37], therefore the restriction to eld- erly patients was removed in the revised 2016 WHO clas- sification [1].
In elderly patients, senescence of immune system in- herent to aging contributes to defective surveillance of EBV, is thought to play a major role in pathogenesis.
Case reports have noted EBV-positive DLBCL concurrent with EBV-positive reactive hyperplasia in a same patient [38], an EBV-positive DLBCL patient with a prior history of mucocutaneous ulcer [39], and EBV-positive reactive hyperplasia that progressed to EBV-positive lymphoma after a couple of years [40]. These case studies support the idea that decreased T cell immunity induced by aging or other causes allows persistence of EBV-infected B cells which continues to proliferate and eventually proceeds to overt lymphoma. Gene expression profiling reveals that antiviral response genes, proinflammatory cyto- kines, and chemokines associated with the innate im- mune response are overexpressed in tumor tissue, in- dicating that a virus induced inflammatory micro- environment plays a role in oncogenesis of EBV-positive DLBCL [41].
The histologic feature is variable ranging from poly-
morphic case with EBV-positive Hodgkin’s lymphoma-
like or T-cell/histiocyte-rich DLBCL-like histologic changes,
to monomorphic lymphoma with necrosis (Fig. 5). The
diagnosis is made through a careful pathological evalua-
tion and detection of EBV-encoded RNA (EBER) in tumor
Fig. 5. (A) EBV-positive diffuse large B cell lymphoma, not otherwise specified. Lymph node biopsy shows large cell lymphoma. (B) CD20.
(C) EBER in situ hybridization.
cells. Although a clear cutoff for positivity has not been defined [1], virtually all tumor cells should be positive for EBV. Tumor cells exhibit mostly type II latency and fewer cases involve type III latency. Most cases exhibit an acti- vated B cell phenotype characterized by NF-kB activa- tion [42]. EBV-positive DLBCL is aggressive disease and prognosis depends on the age of patients. In elderly pa- tients, EBV-positive DLBCL has a lower survival rate than EBV-negative DLBCL, while EBV positivity of DLBCL in young adults is not associated with unfavorable clinical characteristics or worse outcomes [37].
2) EBV-positive mucocutaneous ulcer
EBV positive mucocutaneous ulcer is an indolent con- dition that shows shallow, sharply circumscribed, uni- focal mucosal or cutaneous ulcers that occur in im- munosuppressed patients, including those with age-asso- ciated immunosenescence, iatrogenic immunosuppres- sion, primary immune disorders, and HIV/AIDS-asso- ciated immune deficiencies. The ulcer typically has an indolent course, and spontaneous regression occurs in some cases [1,43-46]. EBV-positive mucocutaneous ulcer can appear in the oral mucosa, skin, or gastrointestinal (GI) tract, but there is no systemic involvement [43].
Patients are mostly adults, with a median age of 77 years (range 42-101 years) reported in one study [43]. The le- sion is usually isolated but can be multiple lesions con- fined to a single anatomic area [46]. Histologically the le- sion is characterized by a polymorphous infiltrate and atypical large B-cell blasts that often exhibit Hodgkin/
Reed-Sternberg (HRS) cell-like morphology.
The B cells in EBV-positive mucocutaneous ulcer show strong CD30 and EBER positivity, and some have reduced
CD20 expression in a background of abundant T cells.
Clonality studies have reported variable results: e.g., slightly more than one-third of cases involve clonal Ig re- arrangement, one-third involve clonal T-cell rearrange- ment, and one-third show a restricted T-cell pattern [43,46]. About a half of cases regressed spontaneously with no treatment and 15% showed relapsing and remit- ting course [43]. The iatrogenic lesions respond to a re- duction in immunosuppressive agents. If only histo- logical changes are considered, EBV-associated mucocu- taneous ulcer may be diagnosed as lymphoma. There- fore, careful differentiation is important for avoiding overdiagnosis and overtreatment.
3) Diffuse large B cell lymphoma associated with chronic inflammation
DLBCL associated with chronic inflammation occurs in the setting of long-standing chronic inflammation, is as- sociated with EBV, and usually involves body cavities or narrow spaces. Pyothorax-associated lymphoma repre- sents the prototype of this type of lymphoma [47].
Pyothorax-associated lymphoma is a non-Hodgkin lym- phoma of an exclusively B-cell phenotype that develops in the pleural cavity of patients after more than 20-year history of pyothorax following an artificial pneumo- thorax for the treatment of pulmonary tuberculosis or tuberculous pleuritis [48,49]. Similar tumors reported in the patients with a chronic osteomyelitis and chronic skin ulcer [50], and knee prosthesis implant [51]. EBV la- tency type III is characteristic.
Chronic inflammation in local sites results in local im-
munodeficiency and provides a favorable environment
for EBV-infected B cells to escape from the host immune
surveillance via their secretion of IL-10 [49,52]. IL-6 pro- duced locally within a chronic pyothorax might also pro- mote the development of pyothorax-associated lympho- ma [53]. Recently, EBV-positive pyothorax-associated lymphoma has been shown to involve expression of CCL17 and CCL22 chemokines, which attract CCR4-expressing regulatory T cells among human peripheral blood mono- nuclear cells [54]. DLBCL associated with chronic in- flammation is responsive to chemotherapy, but the over- all prognosis is poor, as shown by a 5-year survival rate of 21.6% [55].
4) Fibrin-associated diffuse large B cell lymphoma DLBCL that develops in the setting of long-standing chronic inflammation is typically associated with EBV and usually presents as a tumor mass found in body cav- ities, as in pyothorax-associated lymphoma. Another un- usual form of EBV-positive large B-cell lymphoma ap- pears along with fibrin mixed with tissue debris in the wall of cysts, hematomas, hydroceles, coronary aneur- ysms, and cardiac myxomas [56-63]. In contrast to DLBCL associated with chronic inflammation, this lesion does not form a mass itself, but instead microscopic foci of atypical (neoplastic) large lymphoid cells are found within the contents of the cysts, curettage material, or the stroma of atrial myxoma [60]. These lymphoid cells are positive for B-cell markers and have a high Ki-67 la- beling index.
The clinical course of fibrin-associated DLBCL is benign. Most cases, particularly those associated with pseudocysts, behave indolently, may be cured by surgery alone, and may represent a form of EBV-positive LPD rather than lymphoma [64]. Large B cells express LMP1 and EBNA2, belonging to EBV latency type III. As with DLBCL associated with chronic inflammation, the patho- genesis of fibrin-associated DLBCL is thought to be re- lated to decreased immunosurveillance acquired at a lo- cal site [65]. Local immunodeficiency at closed space prevents a cytolytic response to EBV-infected cells and may favor the clonal proliferation of EBV-infected B cells.
5) Lymphomatoid granulomatosis
Lymphomatoid granulomatosis (LYG) is an EBV-pos- itive lymphoproliferative disease with characteristic clin- icopathological findings. Clinically, patients have pulmo- nary involvement that usually comprised of multifocal le- sions with a predilection for the lower lobes [1,66]. Extra- pulmonary sites are commonly involved and include the CNS, liver, skin, and kidney. Mucosal sites including the GI tract and oral mucosa can be involved, although this is uncommon. The spleen, bone marrow, and lymph no- des are usually not involved, and caution should be ex- ercised when diagnosing LYG in a patient with enlarged lymph nodes and splenomegaly. Histologically, the le- sions show polymorphous infiltrate comprising of a vari- able number of large atypical cells admixed with many small T cells with an angiocentric distribution [1,66].
LYG is divided into three grades according to the num- ber of EBV-positive B cells. Grade 1 lesions contain a polymorphous lymphoid infiltrate without cytological atypia and with only infrequent EBV-positive cells (<5/HPF). Grade 2 lesions contain occasional large lym- phoid cells in a polymorphous background with EBV- positive cells (5-20/HPF). Grade 3 lesions show an in- flammatory background with increased EBV-positive cells (>50/HPF). EBER-positive cells can be of various sizes, and numerous small B cells are also positive for EBER. Necrosis is common [66].
Patients typically presents in the fifth decade and there is a male predominance [66]. Patients with under- lying immunodeficiency are at increased risk for LYG.
Most patients show no evidence of underlying known im- munodeficiency however usually manifest reduced im- mune function in EBV surveillance in laboratory analysis [1]. The EBV latency pattern of LYG has been reported as type III latency in 70% of cases examined [66,67].
Differential diagnosis includes other EBV-positive lym-
phoproliferative disease including EBV-positive DLBCL,
NOS. However, a uniform population of atypical EBV-
positive B cells without a polymorphous background is
beyond the spectrum of LYG and should be classified as
diffuse large B-cell lymphoma [22]. The clinical course
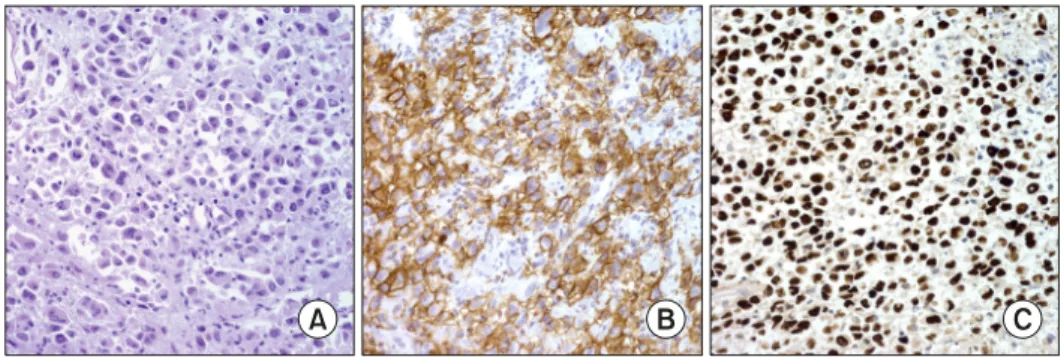
Fig. 6. (A) Extranodal NK/T cell lymphoma. Nasal biopsy shows polymorphic atypical tumor cells. (B) CD3. (C) CD56. (D) EBER in situ hybridization.
ranges from indolent disease to aggressive large B-cell lymphoma. The treatment is selected according to the histological grade and underlying pathobiology, and in- cludes augmentation of the immune response to EBV for low-grade LYG and immunotherapy for high-grade dis- ease [68].
6) Extranodal NK/T cell lymphoma, nasal-type
Extranodal NK/T-cell lymphoma, nasal-type (ENKTL) is defined as a EBV-positive NK or cytotoxic T-cell lym- phoma arising in extranodal sites. The nasal cavity is the prototypic site of involvement. Necrosis of tumor tissue with angiodestructive and angiocentric infiltration of tu- mor cells is characteristic. EBV positivity is observed in virtually all neoplastic cells [1]. At the initial presenta- tion, bone marrow involvement is uncommon. ENKTL accounts for 2.6-12% of non-Hodgkin lymphoma cases in East Asian countries and for 0.5-7.8% in Latin American countries, but <1% of cases in Caucasians [69].
The fact that ENKTL is clustered in East Asia and Latin America suggests that genetic and environmental factors are important in the disease development. Evidence from genetic studies, including mitochondrial DNA and Y chromosome haplotypes, indicates an Asian ancestry of the native population in Latin America, especially in countries located along the Pacific coast [70]. ENKTL af- fects mainly adults, and the median age is 44-50 years.
Its incidence begins to increase in the third decade of life and this form is a major subtype of EBV-positive T/NK-cell
lymphoproliferative disease throughout life in Asia. The disease rarely affects children. In children, NK/T-cell lymphoma may be associated with mosquito bite hyper- sensitivity or systemic CAEBV infection [69].
The typical immunophenotype of ENKTL is CD2+, cy- toplasmic CD3+, CD4−, CD5−, CD8− and CD56+, and involves expression of cytotoxic molecules such as TIA-1, granzyme B, and perforin. While NK-cell marker CD56 is positive in more than 90% of cases, CD16 is usu- ally negative. NK cell receptors such as CD94-NKG2 and KIRs are expressed in 75% and 30% of cases, respectively.
CD30 is expressed in fewer than 50% of ENKTL cases in a subset of tumor cells. PDL-1 is commonly expressed in tumor as well as in background immune cells (Fig. 6) [50, 71].
Tumors of T-cell origin account for 46% of ENKTL cases, and half of T cell ENKTL are T-cell receptor silent [72-74]. Next-generation sequencing studies have identi- fied recurrent mutations shared by patients with ENKTL of NK- and T-cell origin, including mutations involving the RNA helicase gene DDX3X, tumor suppressors, JAK–
STAT pathway molecules, and epigenetic modifiers [75-78].
The overall 5-year survival rates for localized and ad-
vanced ENKTL were reported as 70% and 24%, re-
spectively. Factors indicating a poor prognosis include
nonnasal tumors, advanced stage, high viral load in the
blood, and an immune-silenced tumor microenviron-
ment. Prognostic models based on clinicopathological
parameters and EBV DNA load are useful in the strat-
ification of patients for therapy [71,79,80].
7) Aggressive NK cell leukemia
Aggressive NK cell leukemia (ANKL) is a systemic neo- plastic proliferation of NK cells that is almost always as- sociated with EBV. It is most commonly found in the bone marrow, peripheral blood, liver, and spleen along with pancytopenia [1]. The clinical course is very ag- gressive, and most patients die within a few months of disseminated intravascular coagulation, hemophagocytic syndrome, and multiorgan failure [81]. ANKL is closely associated with EBV, and only 10% of ANKL cases are negative for EBV [82]. ANKL usually presents among young adults or the elderly, and the median age at pre- sentation is 49 years. As for other EBV-positive lympho- mas, the frequency is much higher in Asians than in Caucasians. The high prevalence in Asians is associated with EBV-positive disease and seems to be lower in those with EBV-negative disease [83]. EBV-negative and EBV- positive ANKL patients have similar clinical and patho- logical characteristics, but it is unclear whether the clin- ical outcomes are similar [82,84]. De novo ANKL ac- counts for 80% of cases, and the other 20% evolve from chronic active EBV infection, severe mosquito bite al- lergy, or chronic LPD of NK cells. ANKL arising from CAEBV infection or severe mosquito bite allergy tends to develop more often in younger patients than does de no- vo ANKL [85].
The bone marrow section shows diffuse destructive in- filtration of atypical tumor cells and hemophagocytic histiocytes. The morphology of tumor cells is variable in individual patients and may range from large granular lymphocytes to large pleomorphic cells. Tumor cells ex- press markers that delineate the NK lineage including CD16, CD56, and cCD3. sCD3 and myeloid markers are negative, and the TCR gene is in the germline confi- guration.
ANKL differs from ENKTL in both the clinical findings and the cell lineage of tumor cells. ENKTL involves tu- mors of NK or cytotoxic T cells, whereas ANKL involves tumors of NK cells. ENKTL usually has an extranodal in- volvement such as in the nasal cavity, skin, or GI tract even in cases initially diagnosed in bone marrow biopsy.
ANKL may exhibit similar clinical features as systemic T-cell lymphoma of childhood, but ANKL is rare in chil- dren and involves NK cells, whereas most cases of sys- temic T-cell lymphoma of childhood involve CD8+ cyto- toxic T cells. Genetically, ANKL is characterized by mu- tations of the JAK–STAT pathway, which is also recurrent in ENKTL [78,86,87].
8) Peripheral T cell lymphoma, not otherwise specified, EBV-positive
EBV can infect non-neoplastic B cells or neoplastic T cells in mature T-cell lymphoma. The patient’s prognosis worsens as the number of EBV-positive cells increases.
In this situation, EBV-infected cells account for a very small fraction of infiltrating cells. Apart from such exam- ples, rare nodal T-cell lymphomas in which virtually all tumor cells are EBV positive has been reported in Asia [88-90]. EBV-positive nodal T cell lymphoma is not a separate entity but described as a EBV-positive variant of peripheral T cell lymphoma, not otherwise specified (PTCL, NOS) in the revised 2016 WHO classification.
Median age of patients is 64 years (range, from 25 to 90 years) [89]. Patients have no definite immunodeficiency but are often older adults with a history of other viral in- fections such as HBV and HCV, or diabetes mellitus, which suggests that these patients have an impaired im- mune function that allows viral persistence [89].
Most patients showed adverse clinical features, includ-
ing advanced Ann Arbor stage and high or high/inter-
mediate international prognostic index. Biopsy shows
diffusely proliferating small- medium- to large-sized
atypical pleomorphic or monomorphic tumor cells with
an often centroblastic morphology. Majority of tumor
cells are of cytotoxic αβ -T cell followed by (in decreasing
order of frequency) TCR-silent cytotoxic T cell, and un-
commonly γδ -T cell and NK cell [90,91]. Tumor cells fre-
quently show 14q11.2 loss which correlates with loss of
TCRA loci and upregulation of PDL1 [92]. The tumors
have poor treatment outcomes similar to ANKL, and the
median overall survival is 1.5 months [93].
Conclusion
EBV-positive LPD shows a variety of clinical features and histological findings. To diagnose these accurately, it is important to consider the patient’s clinical findings in detail and to test for EBV for all types of lymphoproli- ferative diseases.
Conflict of Interest Statement
The author has no conflict of interest to declare.
References
1. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th ed. Lyon: IARC Press, 2017.
2. Kimura H, Cohen JI. Chronic active epstein-barr virus dis- ease. Front Immunol 2017;8:1867.
3. Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lym- phoproliferative disease of childhood. Korean J Pathol 2013;47:137-47.
4. Kimura H, Morishima T, Kanegane H, et al. Prognostic fac- tors for chronic active Epstein-Barr virus infection. J Infect Dis 2003;187:527-33.
5. Cohen JI, Jaffe ES, Dale JK, et al. Characterization and treat- ment of chronic active Epstein-Barr virus disease: a 28-year experience in the United States. Blood 2011;117:5835-49.
6. Aoukaty A, Lee IF, Wu J, Tan R. Chronic active Epstein-Barr virus infection associated with low expression of leuko- cyte-associated immunoglobulin-like receptor-1 (LAIR-1) on natural killer cells. J Clin Immunol 2003;23:141-5.
7. Tsuge I, Morishima T, Kimura H, Kuzushima K, Matsuoka H.
Impaired cytotoxic T lymphocyte response to Epstein-Barr virus-infected NK cells in patients with severe chronic active EBV infection. J Med Virol 2001;64:141-8.
8. Okuno Y, Murata T, Sato Y, et al. Defective Epstein-Barr vi- rus in chronic active infection and haematological malignancy.
Nat Microbiol 2019;4:404-13.
9. Yonese I, Sakashita C, Imadome KI, et al. Nationwide survey of systemic chronic active EBV infection in Japan in accord- ance with the new WHO classification. Blood Adv 2020;4:
2918-26.
10. Tatsuno K, Fujiyama T, Matsuoka H, Shimauchi T, Ito T, Tokura Y. Clinical categories of exaggerated skin reactions to mosquito bites and their pathophysiology. J Dermatol Sci 2016;82:145-52.
11. Chung JS, Shin HJ, Lee EY, Cho GJ. Hypersensitivity to mos- quito bites associated with natural killer cell-derived large granular lymphocyte lymphocytosis: a case report in Korea.
Koren J Intern Med 2003;18:50-2.
12. Said J, Smart C. Severe mosquito bite allergy: an unusual EBV(+) NK cell lymphoproliferative disorder. Blood 2019;133:
999.
13. Tokura Y, Ishihara S, Tagawa S, Seo N, Ohshima K, Takigawa M. Hypersensitivity to mosquito bites as the primary clinical manifestation of a juvenile type of Epstein-Barr virus-asso- ciated natural killer cell leukemia/lymphoma. J Am Acad Dermatol 2001;45:569-78.
14. Asada H, Saito-Katsuragi M, Niizeki H, et al. Mosquito sali- vary gland extracts induce EBV-infected NK cell oncogenesis via CD4 T cells in patients with hypersensitivity to mosquito bites. J Invest Dermatol 2005;125:956-61.
15. Tokura Y, Matsuoka H, Koga C, et al. Enhanced T-cell re- sponse to mosquito extracts by NK cells in hypersensitivity to mosquito bites associated with EBV infection and NK cell lymphocytosis. Cancer Sci 2005;96:519-26.
16. Ko Y, Chan J, Quintanilla-Fend L, Jaffe E. Virally associated T-cell and NK cell neoplasms. In: Jaffe E, Arber D, Campo E, Quintanilla-Fend L, Orazi A, editors. Hematopathology.
2nd ed. Philadelphia: Elsevier, 2017.
17. Cho JH, Kim HS, Ko YH, Park CS. Epstein-Barr virus infected natural killer cell lymphoma in a patient with hypersensitivity to mosquito bite. J Infect 2006;52:e173-6.
18. Magaña M, Sangüeza P, Gil-Beristain J, et al. Angiocentric cu- taneous T-cell lymphoma of childhood (hydroa-like lympho- ma): a distinctive type of cutaneous T-cell lymphoma. J Am Acad Dermatol 1998;38:574-9.
19. Ruiz-Maldonado R, Parrilla FM, Orozco-Covarrubias ML, Ridaura C, Tamayo Sanchez L, Duran McKinster C. Edematous, scarring vasculitic panniculitis: a new multisystemic disease with malignant potential. J Am Acad Dermatol 1995;32:37- 44.
20. Cho KH, Lee SH, Kim CW, et al. Epstein-Barr virus-asso- ciated lymphoproliferative lesions presenting as a hydroa vacciniforme-like eruption: an analysis of six cases. Br J Dermatol 2004;151:372-80.
21. Iwatsuki K, Satoh M, Yamamoto T, et al. Pathogenic link be- tween hydroa vacciniforme and Epstein-Barr virus-asso- ciated hematologic disorders. Arch Dermatol 2006;142:587- 95.
22. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed.
Lyon: IARC Press, 2008.
23. Jung SE, Cho KH, Lee MW, Kim YC. Hydroa vacciniforme- like eruption associated with Epstein-Barr Virus infection in an older adult. Ann Dermatol 2015;27:789-91.
24. Long V, Liang MW, Tan SH. Hydroa vacciniforme-like lym- phoproliferative disorder in an elderly Chinese patient and a literature review of adult cases. Int J Dermatol 2018;57:
1283-92.
25. Quintanilla-Martinez L, Fend F. Deciphering hydroa vac- ciniforme. Blood 2019;133:2735-7.
26. Cohen JI, Manoli I, Dowdell K, et al. Hydroa vacciniforme- like lymphoproliferative disorder: an EBV disease with a low risk of systemic illness in whites. Blood 2019;133:2753-64.
27. Iwatsuki K, Xu Z, Ohtsuka M, Kaneko F. Cutaneous lympho-
proliferative disorders associated with Epstein-Barr virus in- fection: a clinical overview. J Dermatol Sci 2000;22:181-95.
28. Cohen JI, Iwatsuki K, Ko YH, et al. Epstein-Barr virus NK and T cell lymphoproliferative disease: report of a 2018 interna- tional meeting. Leuk Lymphoma 2020;61:808-19.
29. Kikuta H, Sakiyama Y, Matsumoto S, et al. Fatal Epstein-Barr virus-associated hemophagocytic syndrome. Blood 1993;82:
3259-64.
30. Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/
chronic EBV infection: a distinct clinicopathologic syndrome.
Blood 2000;96:443-51.
31. Imashuku S, Ueda I, Kusunose S, Shimizu T, Hibi S. Fatal he- mophagocytic lymphohistiocytosis with clonal and granular T cell proliferation in an infant. Acta Haematol 2003;110:
217-9.
32. Lay JD, Tsao CJ, Chen JY, Kadin ME, Su IJ. Upregulation of tumor necrosis factor-alpha gene by Epstein-Barr virus and activation of macrophages in Epstein-Barr virus-infected T cells in the pathogenesis of hemophagocytic syndrome. J Clin Invest 1997;100:1969-79.
33. Coffey AM, Lewis A, Marcogliese AN, et al. A clinicopatho- logic study of the spectrum of systemic forms of EBV-asso- ciated T-cell lymphoproliferative disorders of childhood: a single tertiary care pediatric institution experience in North America. Pediatr Blood Cancer 2019;66:e27798.
34. Smith MC, Cohen DN, Greig B, et al. The ambiguous boun- dary between EBV-related hemophagocytic lymphohistiocy- tosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol 2014;7:5738-49.
35. Imashuku S, Hibi S, Tabata Y, et. al. Outcome of clonal he- mophagocytic lymphohistiocytosis: analysis of 32 cases. Leuk Lymphoma 2000;37:577-84.
36. Beltran BE, Morales D, Quiñones P, Medeiros LJ, Miranda RN, Castillo JJ. EBV-positive diffuse large B-cell lymphoma in young immunocompetent individuals. Clin Lymphoma Myeloma Leuk 2011;11:512-6.
37. Hong JY, Yoon DH, Suh C, et al. EBV-positive diffuse large B-cell lymphoma in young adults: is this a distinct disease entity? Ann Oncol 2015;26:548-55.
38. Gibier JB, Bouchindhomme B, Dubois R, Hivert B, Grardel N, Copin MC. Coexistence of age-related EBV-associated fol- licular hyperplasia and large B-cell EBV+ lymphoma of the elderly. Two distinct features of the same T-cell dysfunction related to senescence? Pathol Res Pract 2017;213:277-80.
39. Daroontum T, Kohno K, Inaguma Y, et al. Epstein-Barr virus (EBV)-positive diffuse large B-cell lymphoma arising in pa- tient with a history of EBV-positive mucocutaneous ulcer and EBV-positive nodal polymorphous B-lymphoproliferative disorder. Pathol Int 2019;69:37-41.
40. Kunitomi A, Hasegawa Y, Asano N, et al. EBV-positive re- active hyperplasia progressed into EBV-positive diffuse large B-cell lymphoma of the elderly over a 6-year period. Intern Med 2018;57:1287-90.
41. Yoon H, Park S, Ju H, et al. Integrated copy number and gene expression profiling analysis of Epstein-Barr virus-positive diffuse large B-cell lymphoma. Genes Chromosomes Cancer
2015;54:383-96.
42. Montes-Moreno S, Odqvist L, Diaz-Perez JA, et al. EBV-pos- itive diffuse large B-cell lymphoma of the elderly is an ag- gressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod Pathol 2012;
25:968-82.
43. Dojcinov SD, Venkataraman G, Raffeld M, Pittaluga S, Jaffe ES. EBV positive mucocutaneous ulcer-a study of 26 cases as- sociated with various sources of immunosuppression. Am J Surg Pathol 2010;34:405-17.
44. Ravi PY, Sigamani E, Jeelani Y, Manipadam MT. Methotrexate- associated Epstein-Barr virus mucocutaneous ulcer: a case report and review of literature. Indian J Pathol Microbiol 2018;61:255-7.
45. Ikeda T, Gion Y, Yoshino T, Sato Y. A review of EBV-positive mucocutaneous ulcers focusing on clinical and pathological aspects. J Clin Exp Hematop 2019;59:64-71.
46. Prieto-Torres L, Eraña I, Gil-Redondo R, et al. The spectrum of EBV-positive mucocutaneous ulcer: a study of 9 cases. Am J Surg Pathol 2019;43:201-10.
47. Pittaluga S, Said J. Virally associated B-cell lymphoprolifer- ative disease. In: Jaffe E, Arber D, Campo E, Harris N, Quintanilla-Martinez L, editors. Hematopathology. 2nd ed.
Philadelphia: Elsevier, 2017.
48. Aozasa K. Pyothorax-associated lymphoma. Int J Hematol 1996;65:9-16.
49. Aozasa K, Takakuwa T, Nakatsuka S. Pyothorax-associated lymphoma: a lymphoma developing in chronic inflammation.
Adv Anat Pathol 2005;12:324-31.
50. Copie-Bergman C, Niedobitek G, Mangham DC, et al. Epstein- Barr virus in B-cell lymphomas associated with chronic sup- purative inflammation. J Pathol 1997;183:287-92.
51. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH.
Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol 2005;29:832-6.
52. Kanno H, Naka N, Yasunaga Y, Aozasa K. Role of an im- munosuppressive cytokine, interleukin-10, in the develop- ment of pyothorax-associated lymphoma. Leukemia 1997;11 Suppl 3:525-6.
53. Kanno H, Yasunaga Y, Iuchi K, et al. Interleukin-6-mediated growth enhancement of cell lines derived from pyothor- ax-associated lymphoma. Lab Invest 1996;75:167-73.
54. Higuchi T, Matsuo K, Hashida Y, et al. Epstein-Barr vi- rus-positive pyothorax-associated lymphoma expresses CCL17 and CCL22 chemokines that attract CCR4-expressing regu- latory T cells. Cancer Lett 2019;453:184-92.
55. Nakatsuka S, Yao M, Hoshida Y, Yamamoto S, Iuchi K, Aozasa K. Pyothorax-associated lymphoma: a review of 106 cases. J Clin Oncol 2002;20:4255-60.
56. Boroumand N, Ly TL, Sonstein J, Medeiros LJ. Microscopic diffuse large B-cell lymphoma (DLBCL) occurring in pseudo- cysts: do these tumors belong to the category of DLBCL asso- ciated with chronic inflammation? Am J Surg Pathol 2012;
36:1074-80.
57. Fujimoto M, Haga H, Okamoto M, et al. EBV-associated dif- fuse large B-cell lymphoma arising in the chest wall with sur-
gical mesh implant. Pathol Int 2008;58:668-71.
58. Garces S, Sriganeshan V. Fibrin-associated diffuse large B- cell lymphoma confined to a cardiac myxoma. Blood 2019;
133:882.
59. Kameda K, Shono T, Takagishi S, et al. Epstein-Barr vi- rus-positive diffuse large B-cell primary central nervous sys- tem lymphoma associated with organized chronic subdural hematoma: a case report and review of the literature. Pathol Int 2015;65:138-43.
60. Loong F, Chan AC, Ho BC, et al. Diffuse large B-cell lympho- ma associated with chronic inflammation as an incidental finding and new clinical scenarios. Mod Pathol 2010;23:493- 501.
61. Yan J, Luo D, Zhang F, et al. Diffuse large B cell lymphoma associated with chronic inflammation arising within atrial myxoma: aggressive histological features but indolent clinical behaviour. Histopathology 2017;71:951-9.
62. Yorita K, Tanaka Y, Hirano K, et al. Fibrin-associated diffuse large B-cell lymphoma arising in a mature cystic teratoma:
a case report. Pathol Int 2019;69:312-4.
63. Zanelli M, Zizzo M, Montanaro M, et al. Fibrin-associated large B-cell lymphoma: first case report within a cerebral ar- tery aneurysm and literature review. BMC Cancer 2019;19:
916.
64. Boyer DF, McKelvie PA, de Leval L, et al. Fibrin-associated EBV-positive large B-cell lymphoma: an indolent neoplasm with features distinct from diffuse large B-cell lymphoma as- sociated with chronic inflammation. Am J Surg Pathol 2017;
41:299-312.
65. King RL, Goodlad JR, Calaminici M, et al. Lymphomas arising in immune-privileged sites: insights into biology, diagnosis, and pathogenesis. Virchows Arch 2020;476:647-65.
66. Song JY, Pittaluga S, Dunleavy K, et al. Lymphomatoid gran- ulomatosis, a single institute experience: pathologic findings and clinical correlations. Am J Surg Pathol 2015;39:141-56.
67. Tanière P, Thivolet-Béjui F, Vitrey D, et al. Lymphomatoid granulomatosis - a report on four cases: evidence for B phe- notype of the tumoral cells. Eur Respir J 1998;12:102-6.
68. Melani C, Jaffe ES, Wilson WH. Pathobiology and treatment of lymphomatoid granulomatosis, a rare EBV-driven disorder.
Blood 2020;135:1344-52.
69. Park S, Ko YH. Epstein-Barr virus-associated T/natural kill- er-cell lymphoproliferative disorders. J Dermatol 2014;41:29- 39.
70. Battaglia V, Grugni V, Perego UA, et al. The first peopling of South America: new evidence from Y-chromosome hap- logroup Q. PLoS One 2013;8:e71390.
71. Cho J, Kim SJ, Park WY, et al. Immune subtyping of extra- nodal NK/T-cell lymphoma: a new biomarker and an im- mune shift during disease progression. Mod Pathol 2020;33:
603-15.
72. Miyata-Takata T, Chuang SS, Takata K, et al. Expression of T-cell receptor signalling pathway components in extranodal NK/T-cell lymphoma. Histopathology 2018;73:1030-8.
73. Hong M, Lee T, Young Kang S, Kim SJ, Kim W, Ko YH. Nasal- type NK/T-cell lymphomas are more frequently T rather than NK lineage based on T-cell receptor gene, RNA, and protein
studies: lineage does not predict clinical behavior. Mod Pathol 2016;29:430-43.
74. Takayama T, Shin S, Kang S, Kim SJ, Kim WS, Ko YH.
Identification of T-cell receptor expression in EBV-positive neoplastic cells in extranodal NK/T-cell lymphoma, na- sal-type, and comparison with T-cell receptor gene re- arrangement by BIOMED-2 assay. Hum Pathol 2018;73:51-8.
75. Peng RJ, Han BW, Cai QQ, et al. Genomic and transcriptomic landscapes of Epstein-Barr virus in extranodal natural killer T-cell lymphoma. Leukemia 2019;33:1451-62.
76. Montes-Mojarro IA, Chen BJ, Ramirez-Ibarguen AF, et al.
Mutational profile and EBV strains of extranodal NK/T-cell lymphoma, nasal type in Latin America. Mod Pathol 2020;
33:781-91.
77. Jiang L, Gu ZH, Yan ZX, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma.
Nat Genet 2015;47:1061-6.
78. Lee S, Park HY, Kang SY, et al. Genetic alterations of JAK/STAT cascade and histone modification in extranodal NK/T-cell lymphoma nasal type. Oncotarget 2015;6:17764- 76.
79. Jung KS, Cho SH, Kim SJ, Ko YH, Kang ES, Kim WS. L-aspar- aginase-based regimens followed by allogeneic hematopoietic stem cell transplantation improve outcomes in aggressive natural killer cell leukemia. J Hematol Oncol 2016;9:41.
80. Kim HS, Kim KH, Kim KH, et al. Whole blood Epstein-Barr virus DNA load as a diagnostic and prognostic surrogate: ex- tranodal natural killer/T-cell lymphoma. Leuk Lymphoma 2009;
50:757-63.
81. Ishida F. Aggressive NK-cell leukemia. Front Pediatr 2018;
6:292.
82. Ko YH, Park S, Kim K, Kim SJ, Kim WS. Aggressive natural killer cell leukemia: is Epstein-Barr virus negativity an in- dicator of a favorable prognosis? Acta Haematol 2008;120:
199-206.
83. Nicolae A, Ganapathi KA, Pham TH, et al. EBV-negative ag- gressive NK-cell leukemia/lymphoma: clinical, pathologic, and genetic features. Am J Surg Pathol 2017;41:67-74.
84. Nicolae A, Pittaluga S, Abdullah S, et al. EBV-positive large B-cell lymphomas in young patients: a nodal lymphoma with evidence for a tolerogenic immune environment. Blood 2015;
126:863-72.
85. Ruskova A, Thula R, Chan G. Aggressive natural killer-cell leukemia: report of five cases and review of the literature.
Leuk Lymphoma 2004;45:2427-38.
86. Dufva O, Kankainen M, Kelkka T, et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat Com- mun 2018;9:1567.
87. Huang L, Liu D, Wang N, et al. Integrated genomic analysis identifies deregulated JAK/STAT-MYC-biosynthesis axis in aggressive NK-cell leukemia. Cell Res 2018;28:172-86.
88. Ha SY, Sung J, Ju H, et al. Epstein-Barr virus-positive nodal peripheral T cell lymphomas: clinicopathologic and gene ex- pression profiling study. Pathol Res Pract 2013;209:448-54.
89. Jeon YK, Kim JH, Sung JY, Han JH, Ko YH; Hematopathology Study Group of the Korean Society of Pathologists. Epstein-
Barr virus-positive nodal T/NK-cell lymphoma: an analysis of 15 cases with distinct clinicopathological features. Hum Pathol 2015;46:981-90.
90. Kato S, Takahashi E, Asano N, et al. Nodal cytotoxic mole- cule (CM)-positive Epstein-Barr virus (EBV)-associated pe- ripheral T cell lymphoma (PTCL): a clinicopathological study of 26 cases. Histopathology 2012;61:186-99.
91. Kato S, Asano N, Miyata-Takata T, et al. T-cell receptor (TCR) phenotype of nodal Epstein-Barr virus (EBV)-positive
cytotoxic T-cell lymphoma (CTL): a clinicopathologic study of 39 cases. Am J Surg Pathol 2015;39:462-71.
92. Ng SB, Chung TH, Kato S, et al. Epstein-Barr virus-associated primary nodal T/NK-cell lymphoma shows a distinct molec- ular signature and copy number changes. Haematologica 2018;103:278-87.
93. Jung KS, Cho SH, Kim SJ, Ko YH, Kim WS. Clinical features and treatment outcome of Epstein-Barr virus-positive nodal T-cell lymphoma. Int J Hematol 2016;104:591-5.