단백질 분자 모델링
김 승 연
한국교통대학교 교양학부 및 융합과학교육대학원
Molecular Modeling for Proteins
Seung-Yeon Kim
School of Liberal Arts and Sciences and Department of Interdisciplinary Science Education, Korea National University of Transportation, Chungju 380-702, Korea
Abstract: 단백질은 생물체의 구성 재료일 뿐만 아니라 생명 활동을 제어하기 때문에 단백질은 생명 현상의 근원이 되는 물질이다. 단백질은 많은 아미노산으로 구성되어 있기 때문에 수많은 원자들을 포함한다. 단백질의 삼차원 구조 는 그 단백질의 기능을 결정하기 때문에, 단백질의 고유한 삼차원 구조가 무엇인지, 그리고 어떠한 과정을 거쳐서 고유 구조로 접히게 되는지 이해하는 것이 매우 중요하다. 선형적인 단백질 일차 구조(유전자 정보로부터 얻어진 아미 노산 서열)로부터 입체적인 단백질 삼차 구조로 접히는 과정을 단백질 폴딩(folding)이라고 부른다. 단백질 폴딩 과정 및 메커니즘은 많은 연구와 전 세계적인 노력에도 불구하고 아직까지 잘 이해되고 있지 않다. 단백질 폴딩이 잘못되면 생명체에 심각한 질병을 초래한다. 단백질의 모든 구성 원자들을 고려하여 이들 사이의 모든 상호작용들을 계산하는 것은 아주 어려운 일이다. 하지만 최근 컴퓨터 기술의 비약적인 발전으로 인해서 근래에는 단백질의 모든 구성 원자들 사이의 모든 상호작용들을 계산하여 단백질의 삼차원 고유 구조 및 폴딩 메커니즘을 규명하려는 연구들이 활발히 시 도되고 있다. 본고에서는 최근 빠르게 발전하고 있는 단백질 분자 모델링에 대해서 소개하였으며 단백질 분자 모델링 에서 이용되는 모든-원자 포텐셜 에너지(all-atom potential energy)에 대해서 설명하였다. 원자 수준에서 단백질의 성 질들을 탐구하기 위해서 가장 많이 사용되는 컴퓨터 계산 방법들인 분자동역학, 몬테카를로 시뮬레이션 방법, 광역 최적화 방법을 간략하게 설명하였다.
Keywords: protein folding, all-atom potential energy, molecular dynamics, Monte Carlo simulation, global optimization
1. 서 론
1)
세포 무게의 거의 대부분을 차지하는 단백질은 생물체의 구성 재료일 뿐만 아니라, 효소, 항체, 호 르몬, 이온 통로(channel) 등이 되어 생명 활동을 제어하기 때문에, 단백질은 생명 현상의 근원이 되는 물질이다. 단백질의 삼차원 구조는 그 단백 질의 기능을 결정하기 때문에, 단백질의 고유한 삼차원 구조가 무엇인지, 그리고 어떠한 과정을 거쳐서 고유 구조(native structure)로 접히게(fol- ding) 되는지 이해하는 것이 매우 중요하다. 폴딩 메카니즘(folding mechanism)과 폴딩 경로(folding
저자 (E-mail: [email protected])
pathway) 연구는 아미노산 서열 정보만으로 출발 한 단백질이 어떤 과정을 통해서 고유의 삼차원 구조를 가지게 되는지를 밝히려는 연구이다. 단백 질이 정상적이지 않은 폴딩 경로를 거쳐서 잘못 폴딩(misfolding)되면 광우병(mad cow disease) 등 과 같은 심각한 질병을 일으킨다. 단백질이 어떠 한 과정을 거쳐 자기 고유의 삼차원 구조로 폴딩 되는지는 실험, 이론, 컴퓨터 시뮬레이션(simula- tion)을 통틀어 아직 아무도 명확하게 밝히지 못한 과제이다.
최근 컴퓨터 기술의 비약적인 발전으로 인해서 근래에는 단백질의 모든 구성 원자들 사이의 모든 상호작용들을 계산하여 단백질의 삼차 구조 및 폴
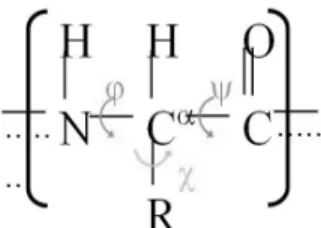
Figure 1. Common structure of one amino acid (N: nitro- gen atom, C: carbon atom, O: oxygen atom, H: hydrogen atom, and R: side chain). Phi, Psi, and Chi are dihedral angles, determining the three-dimensional native structure of a protein.
딩 메커니즘을 규명하려는 연구들이 활발히 시도 되고 있으며 획기적인 연구 결과들이 얻어지고 있 다. 이 글에서는 최근 빠르게 발전하고 있는 단백 질 분자 모델링에 대해서 간단하게 소개하려고 한 다. 다음 절에서는 단백질 구조들에 대해서 간략 하게 설명하였다. 3절에서는 단백질 분자 모델링 에서 이용되는 모든-원자 포텐셜 에너지(all-atom potential energy)에 대해서 설명하였다. 원자 수준 에서 단백질의 성질들을 탐구하기 위해서는 모든- 원자 포텐셜 에너지 외에도 강력한 컴퓨터 계산 방법들이 필요하다. 4절에서 6절까지는 가장 많이 사용되는 컴퓨터 계산 방법들인 분자동역학(4절), 몬테카를로 시뮬레이션 방법(5절), 광역 최적화 방 법(6절)을 아주 짧게 소개하였다.
2. 단백질의 구조
단백질의 기본 구성 요소는 서로 다른 이십 종 류의 아미노산이다. 단백질은 많은 아미노산들이 선형으로 연결되어 만들어진다. 한 아미노산의 척 추(backbone)는 세 개의 원자(질소, 탄소 1, 탄소 2)가 연속적으로 연결되어 이루어져 있고, 이들 원 자들에 각각 수소 1, 수소 2, 산소가 결합되어 있 다. 프롤린(proline)을 제외한 모든 아미노산은 완 전히 똑같은 척추 구조를 가지고 있으며 프롤린의 척추 구조도 다른 아미노산들과 거의 똑같다. 척 추에서 가장 중요한 원자는 중앙에 위치한 탄소 1 인데 탄소 1에는 옆가지(side chain)가 붙어있다.
옆가지의 구성 성분이 아미노산의 종류를 결정한 다. 가장 간단한 아미노산인 글리신(glycine)의 경 우 옆가지는 수소 원자 한 개로만 구성되어 있다.
다음으로 간단한 아미노산인 알라닌(alanine)의 경 우 옆가지는 탄소 원자 한 개와 수소 원자 세 개로 구성되어 있다. 반면 복잡한 아미노산들 중의 하 나인 아르기닌(arginine)의 경우 옆가지는 질소 원 자 세 개, 탄소 원자 네 개 및 수소 원자 열한 개로 구성되어 있다.
유전자를 구성하는 핵산 세 개(코돈-codon-이라 고 불림)가 아미노산의 종류를 결정한다. 즉 특정
유전자의 정보라는 것은 특정 단백질의 아미노산 서열 정보와 동등하다. 특정 단백질의 아미노산 서열 정보는 해당 단백질의 일차 구조(primary structure)라고 불린다. 서로 다른 이십 종류의 아 미노산들은 성질이 서로 크게 다르다. 하지만 아 미노산들을 이들의 성질에 따라 근사적으로 두 부 류-물을 좋아하는 극성 아미노산 부류와 물을 싫 어하는 비극성 아미노산 부류-로 분류할 수 있으 며, 이런 성질 때문에 일차원적인 단백질의 일차 구조는 삼차원적인 고유의(native) 입체 구조[1,2]
를 갖게 된다. 특정 단백질이 삼차원적인 고유 입 체 구조로 접힌 후에 자신의 생물학적 기능들을 수행할 수 있으며 따라서 생명현상이 유지된다.
특정 단백질의 삼차원적인 고유 입체 구조를 해당 단백질의 삼차 구조(tertiary structure)라고 부른다.
대부분 단백질들의 삼차 구조는 공통된 입체 조각 들-알파 나선(alpha helix) 조각과 베타 병풍(beta sheet) 조각-로 구성되어 있다. 이런 공통된 입체 조각들을 단백질의 이차 구조(secondary structure) 라고 부른다. 선형적인 단백질 일차 구조로부터 입체적인 단백질 삼차 구조로 접히는 과정을 단백 질 폴딩(folding)이라고 부른다. 단백질 폴딩 과정 및 메커니즘은 많은 연구와 전 세계적인 노력에도 불구하고 아직까지 잘 이해되고 있지 않다. 단백 질 폴딩이 잘못되면 생명체에 심각한 질병을 초래 한다.
Figure 2. The native tertiary structure of the crambin protein, consisting of 46 amino acids. Its secondary structure includes one alpha-helix and one beta-sheet.
Figure 3. The native structure of the deoxy human hemoglobin, consisting of 574 amino acids. Its secondary structure includes many alpha-helices.
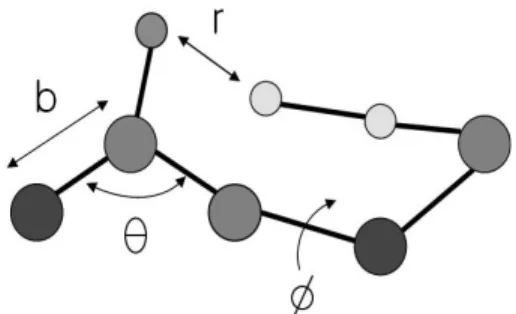
Figure 4. Simple diagram showing the atoms of a protein (b: bond length, theta: bond angle, phi: dihedral angle, and r: distance between two non-bonded atoms).
3. 단백질 분자 모델링에서 이용되는 포텐셜 에너지
단백질은 많은 아미노산으로 구성되어 있기 때 문에 수많은 원자들을 포함한다. 단백질의 모든 구성 원자들을 고려하여 이들 사이의 모든 상호작 용들(interactions)을 계산하는 것은 아주 어려운 일이다. 하지만 최근 컴퓨터 기술의 비약적인 발 전으로 인해서 근래에는 단백질의 모든 구성 원자 들 사이의 모든 상호작용들을 계산하여 단백질의
삼차 구조 및 폴딩 메커니즘을 규명하려는 연구들 이 활발히 시도되고 있으며 획기적인 연구 결과들 이 얻어지고 있다.
단백질의 입체 구조에서 가장 큰 변화를 주는 것은 아미노산 척추에서 정의되는 이면각들 (dihedral angles)이다. 아미노산 척추의 원자인 질 소 원자와 탄소 1 원자 사이에서 이면각 파이가 정의되며 탄소 1 원자와 탄소 2 원자 사이에서 이 면각 프사이가 정의된다. 또한 탄소 2 원자와 다음 아미노산의 질소 원자 사이에서 이면각 오메가가 정의되지만 180도 근처에서 크게 변하지 않기 때 문에 단백질의 삼차 구조에는 큰 영향을 주지는 않는다. 반면 이면각 파이와 프사이는 거의 자유 스럽게 움직일 수 있기 때문에 단백질 삼차 구조 결정에 아주 중요한 역할을 한다.
단백질 분자 모델링[3]에 이용되는 모든-원자 포텐셜 에너지(all-atom potential energy 또는 all- atom force field)는 다양한 에너지 항들로 구성되 어 있다. 첫 번째 고려해야 할 에너지 항은 공유결 합으로 결합된 두 원자들 사이의 결합 길이에 대 한 탄성 에너지이고 두 번째 고려해야 할 에너지 항은 공유결합으로 연결된 세 원자들에 의해 형성 된 결합 각도에 대한 탄성 에너지이다. 이들 두 에 너지 항들은 크게 변하지는 않기 때문에 이들 에 너지 항들을 제외하고 포텐셜 에너지를 계산하는 경우도 있다. 초기 모든-원자 포텐셜 에너지인 ECEPP의 경우 이들 에너지 항들을 포함하지 않 았다. 하지만 최근에는 컴퓨터 성능의 향상으로
Figure 5. An example for the all-atom potential energy of protein. The first and second terms account for the bond length and angle stretching. The third term is related to the energy due to dihedral angles. The last term represents the non-bonded interactions between two atoms i and j, consisting of van der Waals interaction, hydrogen-bonding interaction, and electrostatic interaction.
인해서 현대적인 거의 대부분의 모든-원자 포텐셜 에너지들(AMBER, CHARMM 등)이 이들 에너지 항들을 포함하고 있다. 세 번째 고려해야 할 에너 지 항은 이면각과 관련된 에너지 항이며 가장 필 수적인 에너지 항이다. 이밖에도 단백질 분자 모 델링에 이용되는 모든-원자 포텐셜 에너지에는 비 정상(improper) 이면각과 관련된 에너지 항과 우 레이-브레들리(Urey-Bradley) 에너지 항이 포함된 다. 공유결합으로 연결되지 않은 원자들 사이의 상호작용들도 삼차 구조 결정에 아주 중요한 역할 을 하기 때문에 모든-원자 포텐셜 에너지에 꼭 포 함시켜야 한다. 보통 반데어발스(van der Waals) 상 호작용과 전기(electrostatic) 상호작용을 고려한다.
4. 분자동역학
현재의 실험 방법을 가지고서는 단백질의 일차 구조로부터 단백질의 삼차 구조가 얻어지는 과정 (folding pathways)을 자세하게 조사할 수 없다. 단 백질 폴딩 경로(protein folding pathways)를 자세 하게 조사하기 위해서 다양한 컴퓨터 시뮬레이션 (simulation) 방법들이 사용되고 있다. 이들 방법들 중에서 모든-원자 포텐셜 에너지와 결합된 분자동 역학(molecular dynamics)이 폴딩 경로의 이론적 연구에서 가장 많이 적용된 방법이다. 현재까지 적용된 분자동역학[3] 방법들은 초기치 문제(initial value problem)에 바탕을 두고 있다. 이들 방법들
은 여러 가지 단점들을 가지고 있으며 이들 단점 들 때문에 단백질의 일차 구조로부터 단백질의 삼 차 구조가 얻어지는 과정(folding pathways)을 자 세히 탐구할 수 없다.
기존의 분자동역학 방법들이 가지고 있는 단점 들을 극복하기 위해서, 작용에 바탕을 둔 분자동 역학(action-derived molecular dynamics, ADMD) 도 단백질 폴딩 경로 연구에 적용되고 있다. ADMD 방법[4-7]은 경계치 문제(boundary value problem) 에 바탕을 둔 방법이다. ADMD를 통해서 단백질 삼차 구조들 중에서 가장 기본적인 구조에 해당하 는 알파-나선 구조와 베타-머리띠(hairpin) 구조가 형성되는 과정이 아주 자세하게 조사되고 있다.
현재 ADMD 방법은 좀 더 복잡한 단백질들의 폴 딩 경로 연구에 적용되고 있다.
5. 몬테카를로 시뮬레이션 방법
몬테카를로 시뮬레이션(Monte Carlo simulation) 은 컴퓨터를 이용하여 천문학적으로 큰 수인 상태 수(number of states)를 근사적으로 계산하는 방법 으로서, 다양한 분야에서 애용되고 있다. 가장 잘 알려진 몬테카를로 방법은 메트로폴리스(Metro- polis) 알고리듬으로서 정해진 한 온도에서 가장 큰 공헌을 하는 상태들(states)만을 근사적으로 계 산하는 방법이다. 최근에는 배놑(Bhanot) 알고리 듬, 단일-히스토그램(single-histogram) 방법, 다중- 히스토그램(multiple-histogram) 방법, 다중-정준 (multi-canonical) 방법, 왕-란다우(Wang-Landau) 방 법 등과 같은 강력한 몬테카를로 방법들이 개발되 었다. 이들 방법들은 넓은 온도 구간(또는 모든 온 도 구간)에 걸쳐서 상태 수들을 근사적으로 계산 하는 방법으로서, 단 한 번의 컴퓨터 시뮬레이션 으로 많은 정보를 얻을 수 있다. 이러한 몬테카를 로 시뮬레이션 방법들은 모든-원자 포텐셜 에너지 와 결합되어 이론적인 단백질 연구[8-10]에 큰 공 헌을 하고 있다.
6. 광역 최적화 방법
컴퓨터를 이용하여 주어진 포텐셜에 대해서 광 역 최저점(global minimum)을 찾는 방법을 광역 최적화(global optimization) 방법이라고 부른다.
광역 최적화 방법들 중에서 가장 잘 알려진 방법 은 시늉 담금질(simulated annealing)로서, 이 방법 은 몬테카를로 방법인 메트로폴리스 알고리듬을 이용하는 방법이다. 시늉 담금질 방법보다 더 강 력한 방법으로는 최소 몬테카를로(Monte Carlo with minimization) 방법과 유전 알고리듬(genetic algorithm)이 잘 알려져 있다. 시늉 담금질 방법, 최소 몬테카를로 방법, 유전 알고리듬의 장점들이 적절하게 결합한 광역 최적화 방법들도 개발되어 있다. 이러한 광역 최적화 방법들을 이용하여 단 백질과 같은 복잡한 계들의 특성을 연구할 수 있 고 단백질 삼차 구조 예측 방법들도 개발할 수 있 다[11-17].
복잡한 계에서는 광역 최저점과 국소 최저점들 (local minima)의 분포를 에너지 랜드스케이프 (energy landscape)라고 부른다. 에너지 랜드스케 이프는 복잡계 연구에서 가장 기본적인 개념이며, 해당 시스템의 구조 및 기능을 결정한다. 현재 많 이 연구되고 있는 단백질 폴딩은 아주 거칠거칠한 에너지 랜드스케이프를 가지고 있어서 랜드스케 이프의 성질을 알기 어려우며, 결과적으로 폴딩의 성질을 이해하기가 아주 힘들다. 현재 아주 거칠 거칠한 에너지 랜드스케이프의 성질을 알아내기 위 한 연구가 전 세계적으로 활발하게 진행되고 있다.
단백질의 일차 구조(아미노산 서열)만으로부터 단백질의 삼차 구조를 예측하는 것을 단백질 삼차 구조 예측이라고 한다. 실험 방법을 이용해서 단 백질의 삼차 구조를 결정할 수 있지만 연구 진행 속도가 아주 느리고 비용이 많이 소요되기 때문 에, 많은 국가에서 정부의 주도하에 컴퓨터를 이 용한 구조 예측방법을 개발하려는 연구가 활발히 진행되고 있다. 현재까지 약 일억 개 정도의 유전 자(즉, 단백질의 아미노산 서열)가 밝혀진 반면, 단백질의 삼차 구조는 현재까지 약 만 개 정도밖
에 밝혀지지 않았다. 최근에는 광역 최적화 방법 을 이용하여 모든-원자 포텐셜 에너지에 바탕을 둔 단백질 삼차 구조 예측방법들이 개발되고 있다.
특정 단백질에 붙어서 그 단백질의 역할을 선별 적으로 제어하는 것이 바로 약이다. 따라서 단백 질과 작은 분자(ligand)가 어떤 상호작용으로 서로 붙게 되는지 연구하는 것, 즉 단백질-리간드 도킹 (docking) 문제가 신약개발에 매우 유용하다. 그리 고 단백질들 간의 상호작용을 연구함으로써, 세포 내의 여러 가지 대사 작용을 알아내고 질병의 진 단, 치유에 이용할 수 있다. 광역 최적화 방법과 모든-원자 포텐셜 에너지를 이용하여 단백질-리간 드 도킹 문제와 단백질들 간의 상호작용 연구도 활발히 진행되고 있다.
참 고 문 헌
1. T. E. Creighton, Proteins: Structures and Molecular Properties, 2nd ed. (W. H. Free- man and Company, New York, 1993).
2. A. M. Lesk, Introduction to Protein Architec- ture (Oxford University Press, New York, 2001).
3. T. Schlick, Molecular Modeling and Simula- tion (Springer-Verlag, New York, 2002).
4. I.-H. Lee, S.-Y. Kim, and J. Lee, Chem. Phys.
Lett., 412, 307 (2005).
5. I.-H. Lee, S. Jun, H. Kim, S.-Y. Kim, and J.
Lee, Int. J. Nanotechnology, 3, 334 (2006).
6. I.-H. Lee and S.-Y. Kim, J. Comput. Theor.
Nanosci., 6, 2388 (2009).
7. I.-H. Lee, S.-Y. Kim, and J. Lee, J. Comput.
Chem., 31, 57 (2010).
8. S.-Y. Kim, J. Lee, and J. Lee, J. Chem.
Phys., 120, 8271 (2004).
9. S.-Y. Kim, J. Lee, and J. Lee, Biophys.
Chem., 115, 195 (2005).
10. H. S. Son, S.-Y. Kim, J. Lee, and K.-K. Han, Bioinformatics, 22, 1832 (2006).
11. S.-Y. Kim, S. J. Lee, and J. Lee, J. Chem.
Phys., 119, 10274 (2003).
12. J. Lee, S.-Y. Kim, K. Joo, I. Kim, and J. Lee, Proteins, 56, 704 (2004).
13. K. Lee, C. Czaplewski, S.-Y. Kim, and J.
Lee, J. Comput. Chem., 26, 78 (2005).
14. S.-Y. Kim, S. B. Lee, and J. Lee, Phys. Rev.
E., 72, 011916 (2005).
15. S.-Y. Kim, W. Lee, and J. Lee, J. Chem.
Phys., 125, 194908 (2006).
16. J. Lee, K. Joo, S.-Y. Kim, and J. Lee, J.
Comput. Chem., 29, 2479 (2008).
17. S.-Y. Kim, J. Chem. Phys., 133, 135102 (2010).
김 승 연
1990 연세대학교 학사 1992 연세대학교 석사 1992∼1994 육군화학학교 2000 University of South
Carolina 박사
2000∼2001 Princeton University 연구원 2001∼2005 고등과학원 연구원 2005∼2006 숭실대학교 연구교수 2006∼현재 한국교통대학교 교수
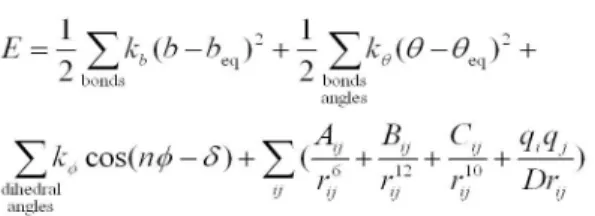
![[기획특집: 신규모델링 기법] 에너지 소재를 위한 분자 동역학과 분자 모델링](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)