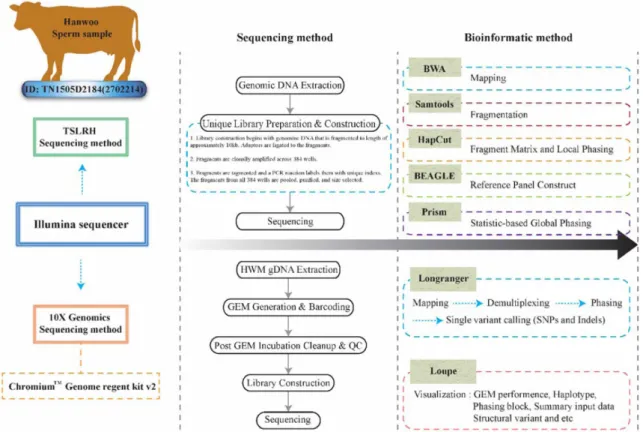
A Comparative Analysis of the Illumina Truseq Synthetic Long-read Haplotyping Sequencing Platform versus the 10X Genomics Chromium Genome Sequencing Platform for Haplotype Phasing and the Identification of Single-nucleotide variants (SNVs) in Hanwoo (Korean Native Cattle)
Woncheoul Park
1, Krishnamoorthy Srikanth
1, Jong-Eun Park
1, Donghyun Shin
2, Haesu Ko
1, Dajeong Lim
1* and In-Cheol Cho
1*
1
Animal Genomics and Bioinformatics Division, National Institute of Animal Science, RDA, Wanju 55365, Korea
2
Departments of Animal Biotechnology, Chonbuk National University, Jeonju 54896, Korea Received July 13, 2018 /Revised November 15, 2018 /Accepted November 19, 2018
In Hanwoo cattle (Korean native cattle), there is a scarcity of comparative analysis papers using high- depth sequencing and haplotype phasing, particularly a comparative analysis of the Truseq Synthetic Long-Read Haplotyping sequencing platform serviced by Illumina (TSLRH) versus the Chromium Genome Sequencing platform serviced by 10X Genomics (10XG). DNA was extracted from the sperm of a Hanwoo breeding bull (ID: TN1505D2184/27214) provided by Hanwoo research canter and used for the generation of sequence data from both the sequencing platforms. We then identified SNVs us- ing an appropriate analysis pipeline tailored for each platform. The TSLRH and 10XG platforms gen- erated a total of 355,208,304 and 1,632,772,004 reads, respectively, corresponding to a Q30 (%) of 89.04%
and 88.60%, respectively, of which 351,992,768(99.09%) and 1,526,641,824(93.50%) were successfully mapped. For the TSLRH and 10XG platforms, the mean depth of the sequencing was 13.04X and 74.3X, the longest phase block was 1,982,706 bp and 1,480,081 bp, the N50 phase block was 57,637 bp and 114,394 bp, the total number of SNVs identified was 4,534,989 and 8,496,813, and the total phased rate was 72.29% and 87.67%, respectively. Moreover, for each chromosome, we identified unique and common SNVs using both sequencing platforms. The number of SNVs was directly proportional to the length of the chromosome. Based on our results, we recommend the use of the 10XG platform for haplotype phasing and SNV identification, as it generated a longer N50 phase block, in addition to a higher mean depth, total number of reads, total number of SNVs, and phase rate, than the TSLRH platform.
Key words : Hanwoo, Haplotype, SNVs, The Chromium Genome Sequencing, TSLRH Sequencing
*Corresponding authors
*Tel : +82-63-238-7306, 7301, Fax : +82-63-238-7347
*E-mail : [email protected] (Dajeong Lim) [email protected] (In-Cheol Cho)
This is an Open-Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Life Science 2019 Vol. 29. No. 1. 1~8 DOI : https://doi.org/10.5352/JLS.2019.29.1.1
서 론
단일염서열변이(SNV, Single Nucleotide Variant)는 단일 염기의 차이를 보이는 변이를 말하며, 단일염기다형성(SNP, Single Nucleotide Polymorphism)과 점 돌연변이(Point mu- tation)을 포함한다. 이러한 단일염기서열변이는 다양한 플랫 폼의 차세대염기서열(NGS, Next Generation Sequence) 데이 터의 분석 결과를 통해 확인 할 수 있다. 또한 차세대 염기서열 데이터가 많아 짐에 따라, 특정 실험설계 및 어플리케이션을 위해 설계된 다양한 기술 및 알고리즘들은 단일염기다형성의
유전자형 분석 수행에 점점 더 널리 이용되고 있다[13]. 현재 많이 사용되고 있는 short-read 시퀀싱 기반으로 단일염기다 형성(SNP, Single Nucleotide Polymorphism)과 염기삽입/결 손(Indel)을 확인하는데 사용된다. 그러나, 이 기술로는 상동 염색체에 대한 염기서열상의 변이를 확인하는데 한계가 있다.
최근 이러한 문제점을 해결하기 위해 생물정보 기반으로 하는
방법들이 만들어 지고 있다. 그 대표적인 예로, 두 쌍의 상동염
색체를 가지는 종에서 보이는 반수체 정보를 이용하는 방법이
다. 이러한 방법들은 연구자들이 DNA 서열과 표현형 사이의
연관성을 이해하는데 많은 도움을 준다[16]. 더욱이 반수체형
은 단일염기다형성 보다 기능적으로 유의미한 자료임과 동시
에 연관불평형 내에서 형질과 연관이 있는 단일염기다형성의
조합이며, 반수체형 자체가 양적형질좌위(QTL, Quantitative
Trait Loci)와 같은 기능을 가지고 있다고 알려져 있다. 반수체
를 분석하기 위한 알고리즘이나 시퀀싱 플랫폼은 현재까지
다양하게 존재한다[15]. 반수체 정보를 확인하기 위하여 현재
가장 대중적인 시퀀싱 플랫폼으로 두 가지 방법이 알려져 있
다. 첫 번째 방법은 Illumina에서 제작된 Truseq Synthetic
싱’ 또는 ‘Genome Phasing’ 이라고도 불린다. 이 방법은 부계 와 모계쪽에서 유전된 대립형질을 염색체 별로 확인해서, 상 동염색체에 대한 유전적 변이를 구별하지 않는 기존의 문제점 을 해결해준다. 또한 혼재된 이형 접합체 분석, 대립형질 특이 적 발현 측정과 변이 링크 확인을 연구하는 연구자들에게도 도움을 준다. 또한 TSLRH Prep Kit를 사용하여 라이브러리를 제작하는 단계를 거친다. 10XG은 Illumina의 short-read 기반 염기서열 데이터로부터 긴 영역의 정보를 산출 할 수 있으며 [4], 1 ng의 유전체 DNA보다 적은 양으로 양질의 라이브러리 를 제작할 수 있다. 또한 10X Genomics Prep Kit을 사용하여 라이브러리 제작이 가능하다[19]. 또한 여기에서 산출 된 데이 터를 분석하기 위한 프로그램은 자체적으로 제작해서 사용하 고 있으며(단계화: LongRanger, 반수체형 단계화 및 구조 변 이의 시각화: Loupe) 다른 많은 논문에서 사용되고 있다[12, 17]. 이 두 가지 시퀀싱 플랫폼은 가격대비 효율이 뛰어나다고 잘 알려져 있다.
한우는 한국 고유 토착 소 품종으로서 한국 내의 소비자에 게 매우 선호도가 높은 고기이다. 왜냐하면 소비자들은 수입 소고기 보다 한우 고기가 더 풍미와 육즙이 좋다고 믿고 있기 때문이다[5, 6]. 이러한 선호도로 인해 한국 내에 한우에 대한 관심과 연구는 많은 분야에서 이루어 지고 있는 실정이다. 그 예로, 유전자 마커, 소수의 양적형질좌위, 유전자에 대한 형질 예측, 고밀도 단일염기다형성 칩을 이용한 유전체 선발, 전장 유전체연관분석(GWAS, Genome-Wide Association Study), 전사체 데이터를 이용한 차등발현유전자 발견 등의 연구가 과거에서 현재까지 이루어 지고 있다[7, 9-11, 18]. 하지만 현재 까지 반수체형 확보는 상용 칩(Illumina, Affymetrix, GenSeek 등)을 기반으로 저밀도와 고밀도 SNP chip을 활용하거나 형질 과 연관된 유전자 영역 내의 단일염기다형성 정보의 조합으로 구성하는 게 전부였다. 그러나 지금까지의 연구결과를 이용한 활용 완성도 및 현장 적용이 높지가 않다.
본 연구는 반수체형 및 단계적 정보를 이용한 최신의 시퀀 스 플랫폼인 TSLRH 그리고 10XG을 이용하여 한국 단일 소 품종인 한우의 형질 예측 및 반수체형 지도를 구축하기 위해 최적화된 시퀀스 플랫폼을 제안하는 것이 본 연구의 목적이 다. 뿐만 아니라, 본 연구에서 분석된 한우 반수체형 정보를 기반으로 추후 한우 집단 유전적 특성을 규명하는 연구에 기 초자료로 활용될 것이라 사료된다.
재료 및 방법
실험 재료
한우 연구소에서 보유하고 있는 후보종모우 중에 자손의
생산하였다(Fig. 1).
DNA 추출 및 차세대 염기서열 자료 생산
동결 정액이 담긴 스트로를 잘라서 1.5 ml 튜브에 담그고, PBS ph7.2 (1X)를 1 ml을 넣고 볼텍싱(vortexing)을 1분 이상 하였다. 원심분리 후 상층액을 제거한다. 이러한 일련의 과정 을 5회 반복하였다. 추출 버퍼(buffer) 250 μl, 10% SDS 35 μl, 0.5 M DTT 30 μl, Protein K (20 mg) 25 μl를 분주 후 볼텍싱 하고 65℃ 항온수조에서 3시간 이상 보관하여 녹인다. 이 후 냉동실에 15분간 얼린 후 상온에 방치 하는데 이를 3회 반복한 다. 5 M Nacl 120 μl를 넣고 볼텍싱, 원심분리 후 새로운 튜브 에 상층액을 옮긴다. 100% Etoh (Ethanol)을 500 μl 주입 후 위아래로 섞어 주고 원심분리 시킨 후 상층액을 제거한다. 70%
Etoh 500 μl를 주입하고 위아래로 섞어 주고 원심분리 한 후 상층액을 제거한다. 마지막으로 Air dry gn D.W. 200~300 μl 을 넣고 냉장 및 냉동 보관한다.
TSLRH 분석 순서로는 DNA 추출, 라이브러리 제작 및 시 퀀싱 자료 생산단계로 진행이 된다. 그 중 라이브러리 제작이 특이한데 조금 더 세밀하게 설명하면 총 3단계로 나뉘어 진다.
첫 번째 단계에서 수집 된 DNA를 약 10 kpb의 파편으로 (fragment) 절단 한다. 두 번째 단계로 파편들을 희석한 후 384 개의 웰(wells) 각각에 3,000개의 파편들을 주입해준다. 세 번 째 단계에서 Long-range PCR을 통해서 각각의 well 단편들을 증폭시키고 다시 단편으로 절단하여 바코드 작업이 진행된다.
그후 시퀀싱 데이터 생산과정을 거친다[8]. 10X Genomics
(https://www.10xgenomics.com/)에서의 10XG 방법은 크게
5단계로 나뉘어 진다. 첫 번째로 고분자량(high molecular
weight) gDNA 추출과정, 두 번째로 GEM 발생 및 바코딩
(generation and barcoding), 세 번째 과정은 발생된 GEM 세
척 및 품질 조정(cleanup and QC), 네 번째 과정은 라이브러리
제작, 마지막으로 시퀀싱 과정을 거친다. 본 연구에서는 자체
적으로 정자에서 DNA 추출을 하였기 때문에 첫 번째 단계는
생략을 하고 두 번째 단계부터 시작하였다. 두 번째 단계인
GEM 발생 및 바코딩은 또 크게 6단계, 추출된 고분자량
gDNA 정량화 삽입, 샘플 마스터 믹스 준비, chromium
TM지
놈 칩 로딩, chromium 컨트롤러 작동, GEMs 운송 및 GEM
등온 배양 순으로 진행된다. 세 번째 단계인 발생된 GEM 세척
및 품질 조정에서는 3단계 즉, Silane DynaBeads, SPRIselect
및 quality control 순으로 진행된다. 네 번째 단계인 라이브러
리 제작에서는 총 7단계, 말단 보정(End repair), A-tailing, 아
답터 결합, 결합물 세척, 샘플 인덱스 PCR, 샘플 인덱스 후
양쪽 사이즈 선택(SPRIselect), 라이브러리 제작 후 quality
control을 거친 후 정량화 단계로 진행된다. 마지막 단계인 시
Fig. 1. The graphical scheme of the experimental design, sequencing and bioinformatics method.
퀀싱은 HiSeqX 플랫폼을 사용하여 Paired-end 방법, 염기서 열 길이는 151 bp, 한우 참조서열 대비 약 40배수로 시퀀싱을 진행 하였다(Fig. 1) [19].
차세대 염기서열 분석 및 비교 분석
TSLRH 방법으로 생산된 차세대 염기서열의 분석과정은 총 7 단계로 구성이 되어 있으며, 분석 방법은 다음과 같은 순서 로 이루어 진다. 1) bwa v0.7.10 버전의 mem 알고리즘으로 맵핑. 2) samtools (v0.0.19) targetcut으로 fragment화. 3) HapCut (v0.7) 내의 extractHAIRS 툴을 이용하여 fragment 매트릭스. 4) 15 kb 이상의 유전체 영역은 키메릭 서열로 가정 하여 제거. 5) HapCut (v0.7)로 local phasing을 수행. 6) 축산 과학원의 한우 136두의 전사체(mRNA-seq) 데이터를 사용하 여 BEAGLE 프로그램으로 참조 패널을 구축. 7) Prism 프로그 램으로 6번째 분석 결과의 해당 부분의 statistic-based global phasing을 수행하였다[8]. 10XG에서 생산된 차세대 염기서열 데이터의 분석 과정은 크게 2가지 프로그램을 이용하며, 10X Genomics에서 제공하는 Long Ranger (v.2.1.6) 프로그램과 Loupe genome browser (v.2.1.1) 이용하여 분석하였다. 첫 번 째 Long Ranger의 분석 순서는 다음과 같다. 1) 샘플의 역다중 화(demultiplexing). 2) 바코드 처리. 3) 참조 유전체(UMD_
3.1/BosTau6:ftp://hgdownload.soe.ucsc.edu/goldenPath/
bosTau6/bigZips/bosTau6.fa.gz)에 생산된 시퀀싱 데이터를
맵핑. 4) 퀄리티 조정. 5) 변이 추출(variant calling). 6) 페이징 (phasing). 7) 구조 변이 추출(structural variant calling) 순서 로 분석이 이루어진다. 두 번째로, Loupe genome browser는 Long Ranger의 결과 파일을 이용하여 시각화 해주는 프로그 램으로써 haplotype, phasing block, 구조 변이 등을 시각화 해 주며 전체적인 수치를 요약해서 보여준다. 최종적으로 TSLRH 과 10XG 방법으로 생산된 차세대염기서열 데이터를 비교 분석 하기 위해 vcftools (v.0.1.12a)을 이용하여 분석하였다(Fig. 1) [2].
결과 및 고찰
시퀀싱 및 기초 분석 비교 결과
한우 대표 종모우의 정액 샘플의(개체 식별 번호: 2702214) 시퀀싱 및 기초 분석은 Table 1으로 확인할 수 있으며 이러한 결과를 도출 하기 위한 일련의 과정들은 Fig. 1을 통해 확인 할 수 있다. 우선 TSLRH 결과 총 염기서열 개수는 355,208,304 개 이며, 35,876,038,704 bp에 해당된다. GC 서열 개수는 14,443,143,551개였으며 GC contents 는 40.26%였다. 더욱이 Q20 이상은 95.66%, Q30 이상은 89.04%인 것을 확인하였다.
이 후 시퀀싱 결과로 생성된 데이터를 소의 참조 서열에 맵핑
한 결과, 참조 유전체에 맵핑이 된 리드 개수는 351,992,768개
였으며 맵핑율은 99.09%였다. 이들 중 쌍으로 제대로 맵핑이
된 리드는 개수는 33,920,746개 였으며 맵핑율은 95.13% 임을
Sequencing platform
10X Genomics The Chromium
Genome
Illumina TSLRH
Total reads Mapped Reads Mapping rate (%) Q20 (%)
Q30 (%) GC Contents (%) Mean depth
Genome Coverage(1X) Longest Phase Block N50 Phase Block Price
1,632,772,004 1,526,641,824
93.50%
89.53%
88.60%
45.86%
74.3X 99.77%
2,490,347 114,853
Low
355,208,304 351,992,768 99.09%
95.66%
89.04%
40.26%
13.04X 98.04%
1,982,706 57,637
High
확인하였다. 또한 평균 리드의 밀도(Mean Depth)는 13.04X 정도이며 전체 리드가 유전체 전체를 차지는 하는 영역 비율 은(Genome Coverage: 1X) 98.04% 였다. 페이징 분석 결과 가 장 긴 페이즈 블락은 1,982,706bp 였으며 N50의 페이즈 블락은 57,637 bp였다. 10XG 결과 전체 리드의 개수는 1,632,772,004 개 였으며, 이 전체 리드들의 총 베이스는 123,274,286,302였다.
GC contents는 45.86% 였으며, Q20 이상은 89.53%, Q30 이상 은 80.36%임을 확인하였다. TSLRH와 마찬 기지로 맵핑한 결 과, 참조 유전체에 맵핑이 된 리드 수는 1,526,641,824개 였다.
맵핑율은 93.5% 로써 TSLRH의 맵핑율과 비슷한 수치를 보여 주고 있다. 또한 평균 리드 밀도는 74.3X 정도이며 유전체 범위 율은 99.77% 였다. 다음으로 단계 페이징 분석 결과 가장 긴 phase block은 2,490,347 bp였으며 N50의 페이즈 블락은 114,853 bp였다(Table. 1). 이와 같은 결과는 아프리카 야생 개 에서의 10XG 결과와 유사하며, 이는 같은 포유 동물에서 비슷 한 결과를 보여 줌으로써 데이터 분석에는 문제가 없음을 확 인할 수 있다[1].
두 가지 시퀀싱 및 기초 분석 결과를 비교해 보면, 우선 GC Contents는 10XG가 조금 더 높은 것을 확인 할 수 있었으나 두 시퀀싱 플랫폼 모두다 포유류에서 보이는 양상과 비슷하 며, 시퀀싱 방법에 따라 GC Contents는 다르나 두 시퀀싱 모두 소 유전체에서 보이는 GC Contents 양상과 비슷한 수치를 보 인다[3, 14]. Q20과 Q30은 TSLRH이 더욱 높은 수치를 보였으 나 Q30이 90% 이상의 고품질은 아니었다. 하지만 Q30이 80%
이상이므로 추후 분석을 하는 데는 문제가 없다고 할 수 있겠 다. 맵핑율은 두 가지 시퀀싱 플랫폼 모두다 90% 이상으로 마찬가지로 추후 분석을 하는 데에는 문제가 없다고 할 수 있다. 여기까지 결과를 비교하면 두 가지 시퀀싱 플랫폼 모두 신뢰할 수 있는 양질의 시퀀싱 데이터를 생산하는 것을 알 수 있다. 조금 더 세부적인 부분을 살펴보면 전체 리드 개수,
것을 확인 할 수 있었다(Table 1).
단일염기서열변이 비교 분석
단일염기서열변이 분석은 각각의 시퀀싱 플랫폼에 따라 각 기 다른 프로그램을 사용해서 발굴을 하였다(Fig. 1). 그 결과, TSLRH으로 발굴한 전체 단일염기다형성의 개수는 4,061,597 개 였다. 그리고 이 단일염기다형성 중 동형접합체(Homozy- gous)의 개수는 219,582개로 비율은 5.41%며 이형접합체(He- terozygous)의 개수는 3,842,015개로 그 비율은 94.59%로 구성 되어 있었다. 더욱이 이 이형접합체에서 페이징이 된 이형접 합체의 개수는 2,666,097개로 그 비율은 69.39%이었다. 전체 Indels의 개수는 473,392개 이었으며, 이 전체 Indels 중에 동형 접합체의 개수는 43,937개 였으며, 그 비율은 9.28%였다. 또한 이형접합체의 개수는 429,455개며 그 비율은 90.72%였다. 그 리고 이 Indels의 이형접합체에서 페이징이 된 이형접합체의 개수는 322,914개며 그 비율은 75.19%였다. 최종적으로 TSLRH 에서 페이징 비율은 72.29%인 것으로 확인되었다. 10XG으로 발굴한 전체 단일염기다형성의 개수는 7,548,893개였다. 이 단 일염기다형성 중 동형접합체의 개수는 2,763,109개로 비율은 36.6%였다. 이형접합체의 개수는 4,785,784개로 그 비율은 63.40%로 구성되어 있었다. 전체 이형접합체에서 페이징이 된 이형 접합체의 개수는 4,580,909개이며 그 비율은 95.72%였다.
전체 Indels의 개수는 947,920개 였으며, 전체 Indels 중에 동형
접합체의 개수는 453,457개였고, 그 비율은 47.84%였다. 또한
이형접합체의 개수는 494,463개였으며, 그 비율은 52.16%였
다. Indels의 이형접합체에서 페이징이 된 이형접합체의 개수
는 393,733개며 그 비율은 79.63%였다. 최종적으로 10XG의
페이징 비율은 87.67%인 것을 확인 하였다(Table 2). 이 결과를
바탕으로, 10XG이 TSLRH 보다 더 많은 단일염기 다형성과
Indels을 발굴할 수 있으며, 더 높은 비율로 페이징을 한다는
것을 확인하였다. 각각의 시퀀싱 플랫폼의 단일염기서열변이
를 비교하기 위해 염색체 별로 분포가 어떻게 되어 있는지
확인하였다. 그 결과, 염색체 길이와 단일염기서열 변이의 분
포는 정비례하는 결과를 보여 주었다. 각각의 플랫폼에서 특
별한 단일염기다형성과 Indels이 존재하며 두 가지 시퀀싱 플
랫폼에서 서로 공통으로 가지는 단일염기다형성과 Indels을
확인할 수 있었다(Fig. 2). 이 공통적인 단일염기서열변이는
대부분의 염색체에서 TSLRH의 단일염기서열변이의 대략 절
반 정도 공통적으로 존재하였다. 또한, 발굴한 단일염기서열
변이가 기존에 존재하는 변이인지 확인하기 위해 참조 단일염
기서열변이의 정보를 이용하여 확인하였다(Table S1). 그 결
과, 10XG과 TSLRH의 전체 단일염기다형성에서 90% 정도가
기존에 알려져 있었고 10% 정도만이 알려지지 않은 단일염기
Table 2. Summary of the number of SNVs and the rate of type SNVs and phasing result from The Chromium Genome of 10X Genomics and TSLRH of Illumina sequencing data
10X Genomics The Chromium Genome Illumina TSLRH
SNPs Indels SNPs Indels
Total Homozygous (%) Heterozygous (%) Phased Heterozygous (%)
7,548,893 2,763,109(36.60%) 4,785,784(63.40%) 4,580,909(95.72%)
947,920 453,457(47.84%) 494,463(52.16%) 393,733(79.63%)
4,061,597 219,582(5.41%) 3,842,015(94.59%) 2,666,097(69.39%)
473,392 43,937(9.28%) 429,455(90.72%) 322,914(75.19%)
Total Phased rate 87.67% 72.29%
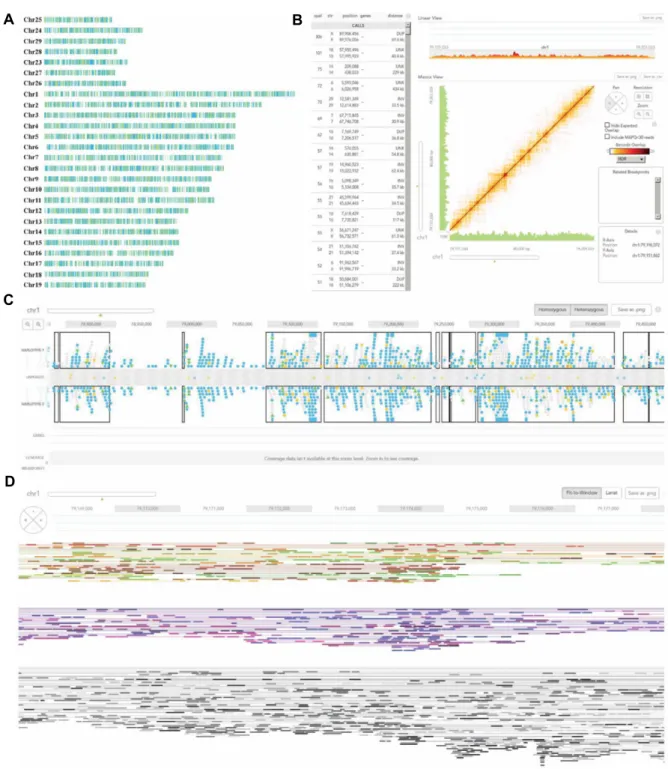
A
B
Fig. 2. Histogram of the number of SNVs from TSLRH of Illumina and The Chromium Genome of 10X Genomics: (A) Histogram of the number of SNPs of TSLRH, 10X and common. (B) Histogram of the number of Indels of TSLRH, 10X and common.
다형성 이었다. 그러나 Indels은 10XG가 77%, TSLRH는 34%
로 10XG가 높게 보이지만 알려지지 않은 Indels의 개수에서는 큰 차이를 보이지 않았다. 이와 같은 결과들을 통해 10XG과 TSLRH은 서로 상호보완적인 역할을 할 수 있다고 판단된다.
하지만 단일염기서열변이를 발굴하는 목적으로는 10XG이 더 적합하다고 사료된다.
본 연구의 결과를 통해 비용의 절감, 많은 단일염기서열변 이 발굴, 반수체의 길이, 페이징 비율 등이 TSLRH보다 상대적 으로 좋은 10XG가 한국의 단일 소 품종인 한우의 반수체형 지도 작성 및 형질 예측 분석에 필요한 시퀀싱 플랫폼으로 적당할 것이라고 사료 된다. 다만 예산이 풍부할 때에는 상호
보완적인 부분이 있는 두 시퀀싱 플랫폼을 같이 쓰는 것이
더 좋은 방법이라고 사료된다. 여기서 상호보완적인 부분은
크게 두 가지로 나뉘어 진다. 첫 번째로 두 플랫폼에서 같은
영역의 단일염기서열변이도 발굴 할 수 있지만 서로 다른 영
역의 단일염기서열변이들도 발굴 하기 때문에 두 시퀀싱 플랫
폼을 다 사용함으로써 더 많은 영역의 단일염기서열 변이들을
발굴 수 있다는 것이다. 두 번째로 양쪽 모든 시퀀싱 플랫폼에
서 발굴된 단일염기서열변이들은 두 플랫폼 모두에서 검증된
단일염기서열변이 이기 때문에 높은 신뢰 값을 가진다고 할
수 있겠다. 또한 추후 한우 맞춤형 칩을 제작할 때 여기서 발굴
한 높은 신뢰 값을 가지는 단일염기서열변이를 바탕으로 제작
C
D
Fig. 3. Visualization of phasing block, haplotype, structural variant and mapped read from The Chromium Genome of 10X Genomics by using Loupe program: (A) Phasing block of each chromosome. (B) Structural variant. (C) Haplotype. (D) Mapped read.
을 하면 정확도가 높은 칩을 제작할 수 있다는 것이다. 또한 본 연구와 같이 반수체형 분석을 위한 시퀀싱 플랫폼을 비교 한 논문은 아직까지 많이 보고 된 바가 없다. 따라서 본 연구는 앞으로 반수체형 분석을 하는 연구자들이 어떠한 시퀀싱 플랫 폼을 따라야 할 지, 예산에 맞는 시퀀싱 플랫폼은 무엇인지를 알려주는 선행 연구라고 사료된다.
감사의 글
본 연구는 농촌진흥청 연구사업(PJ01251901) 연구비를 지 원받아 수행하였으며, 연구비 지원에 감사 드립니다.
References