DOI 10.17480/psk.2017.61.4.216
반경험적 모델계산에 의한 플라본과 플라본유도체의 물성계산
박경래 · 장하영# 충남대학교 약학대학
(Received July 19, 2017; Revised August 20, 2017; Accepted August 26, 2017)
Atomic Charges of Flavone Derivatives by Semiempirical Model Calculation
Kyung Lae Park and Ha Young Jang#
College of Pharmacy, Chungnam National University, Daejeon 34134, Korea
Abstract — For the prediction of atomic properties of some flavone derivatives, semiempirical quantum mechanical cal- culation was performed using the structures from the protein data bank. The structures were geometrically optimized in the framework of molecular mechanics and the atomic charges were calculated with the AM1 method. These virtual com- pounds with explicit atomic charges shall be used for the prediction of molecular interactions in the complex with various receptors, which are known as bioactive proteins. The results showed consistent values with partial atomic charges of con- stituents in protein, adopted in the main molecular mechanics forcefield.
Keywords flavonoids, partial charge, semiempirical method, molecular mechanics
플라보노이드는 플라본(C15H10O2)을 기본 골격으로 하는 화합 물로 지금까지 천연물에서 발견된 플라보노이드는 4,000종 이상 에 이르고 있다.1)주로 과일, 채소 및 녹차류에 풍부하게 존재하 고2)많은 연구에서 플라보노이드는 항산화, 항알레르기, 항바이 러스, 항염증, 혈관확장 등의 생물학적 작용이 있음을 보여주고 있으며 이중 가장 흥미를 끄는 작용은 항산화 효과이다.3)
실제로 자연계에서 발견된 많은 플라보노이드가 특정 단백질 수용체와 일종의 복합체(complex)를 이루고 있으며 이러한 결합 의 본질은 형태가 조금씩 다르기는 하여도 일종의 정전기적 인 력이 될 수밖에 없으며 이러한 정전기 인력의 주요 인자는 각 구 성원자의 부분전하의 형태로 구현될 수 있다. 각 원자의 부분전 하는 수용체 단백질과 플라보노이드의 결합에 직접 관여하기도 하지만 결합 후 2차적인 유도인력에 영향을 끼치기도 한다.
일반적으로 이러한 생체분자들 간의 상호작용 계산에 이용되 는 분자역학적 알고리즘(Molecular Mechanics)은 고전적 뉴톤역
학에 기반을 두고 있지만, 생체분자들 간의 상호작용에 직접적 으로 연관된 분자내 원자의 전하는 양자역학적 개념이어서 분자 역학자체로는 계산되거나 예측될 수 없다. 이러한 경우에 양자 역학적 방법으로 계산된 원자들의 전하를 하나의 고유치 상수로 입력하여 분자역학에서 정전기적 인력의 정량적 계산에 이용하 는 방법을 쓴다.
기존에 존재하는 분자역학적 프로그램의 알고리즘에는 생체분 자에 존재하는 원자의 형태에 맞게 미리 계산되고 정의된 원자 의 부분전하를 내적 데이터베이스의 형태로 저장되어 있으며, 학 계에 발표된 여러 프로그램에는 알고리즘의 개발자에 따라 약간 의 차이가 있는 부분전하가 사용되고 있다. 어느 방법이든 단백 질과 핵산에 존재하는 표준원자들의 결합형태에 따라 50~70가 지의 부분전하가 정의되어 자체의 힘장(force field)에 사용되고 있다. 그러나 자연계에는 일정한 표준원자 이외에 부분적으로 변 이된 아미노산과 원자들이 존재하며, 모든 가능한 변이 형태를 만능적으로 포함하는 힘장을 만들 수는 없으며 특히 생체분자에 리간드로 작용하는 자연물질 또는 인공적으로 합성된 약물들의 가짓수는 무한에 가까우므로 이들 원자 형태들은 어떠한 힘장의 분류에도 포함되지 않은 경우가 대부분이다. 이러한 리간드와 수 용체간의 상호작용의 계산을 위해서는 기존의 힘장에서 찾을 수
#
Corresponding Author Ha Young Jang
College of Pharmacy, Chungnam National University, Daejeon 34134, Korea
Tel.: 042-821-5938 Fax.: 042-821-6566 E-mail: [email protected]
Short Report
종설없는 원자전하를 별도로 계산해서 입력해 줘야 한다. 또한 단백 질이나 핵산의 변성 또는 변이에 의하여 새로운 원자전하의 설 정이 필요한 경우에는 다른 생체분자들의 전하 값과 같은 맥락 의 전하를 새로이 계산하여 입력해 줘야하는 것이다.
과거엔 실험으로만 가능하였던 원자, 분자의 반응이 지금은 컴 퓨터 모의실험을 통하여 일정 부분 예측할 수 있게 되었다. 이러 한 예측이 가능했던 이유는 컴퓨터 연산 속도의 향상과 다양한 소프트웨어도구의 개발의 결과이며 수퍼컴퓨터로만 가능했던 복 잡한 연산이 지금은 개인용 데스크탑 컴퓨터에서도 상당한 수준 의 계산이 가능하게 된 것이다.
원자 또는 분자수준에서의 계산은 크게 양자역학과 분자역학 적 방식으로 구분되고 양자역학은 다시 ab initio 와 반경험적 (semiempirical)으로 나뉜다. 본질적 측면에서 양자역학 방식 중 ab initio 방법이 이론적 정당성을 제시하지만 컴퓨터 용량의 한 계와 기존의 계산된 아미노산과 핵산들의 전하가 대부분 반경험 적 방법에 의한 수치를 기반으로 개발되고 최적화 되어왔기 때 문에 단백질과의 복합제의 경우에도 유사한 방법과 절차에 따라
계산되는 전하값을 사용하는 것이 일관성있고 논리적으로도 합 당할 것이다.
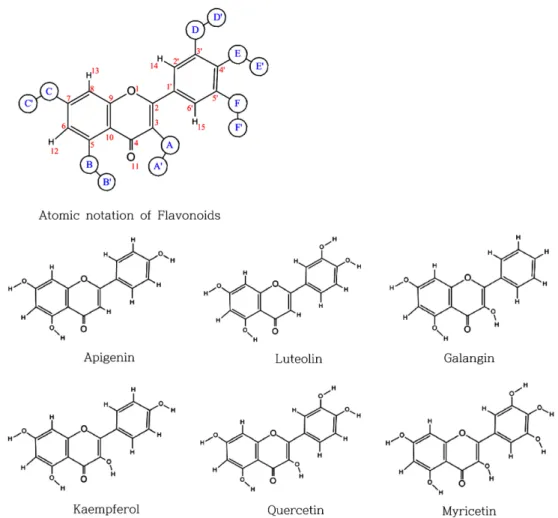
본 연구에서는 반경험적 계산방법인 AM1알고리즘4)을 사용하 여 자연계에서 흔히 발견되는 플라보노이드 중 수용체와 결합한 상태의 x-선 결정구조가 알려져 있는 Flavone, Apigenin, Luteolin, Galangin, Kaempferol, Quercetin 과 Myricetin 등에 대하여 각각 의 원자전하를 계산하였다. 이들은 모두 평면구조를 이루며 순수 탄화수소인 1,4-dihydro-2-phenylnaphthalene (C16H14)과 거의 같은 형태구조를 갖는다. 이러한 플라보노이드 화합물들의 전자 구조적 물성을 조사하기 위하여 2-phenylnaphthalene의 골격으 로부터 출발하여 플라보노이드 화합물로 유도되는 구조를 계산 하여 각 원자의 전하의 변화를 추적하였다.
실험방법 (Experimental Methods)
이 연구의 대상인 플라본 유도체의 구조는 단백질은행 (RCSB Protein Data Bank)5)에 공개된 플라보노이드-단백질 복합체의
Fig. 1 − Atomic numbering of flavonoid derivatives: A, B, C, D and E can be H or O and A’, B’, C’, D’ and E’ can be H, where Flavone has
all hydrogens at position A, B, C, D and E.
구조로부터 내려받은 구조를 바탕으로 사용하였으며 분자내 원 자의 번호는 Fig. 1과 같이 표준 번호체계대로 작성하였다. 초기 분자 편집에는 분자역학적 MM+힘장6)에 의거하여 원자가가 채 워지지 않은 C와 O원자에 대하여 수소원자를 삽입하였다. 플라 본과 그 유도체들의 구조는 원래 두 방향족 고리사이에 여분의 이중결합과 카르보닐결합으로 양쪽의 방향족 고리가 연속적으로 공액화(πconjugation) 되어 있다. 따라서 플라본 유도체들의 전
체적인 형태구조는 거의 평면구조를 벗어날 수 없는 구조이다.
이러한 형태구조에서는 분자의 3차원구조의 기하학적 최적화에 의한 구조의 안정화는 큰 의미를 갖지 않는다. 위의 분자역학적 절차에 따라 탄소와 산소원자의 빈자리에 수소를 채워 넣은 구 조에 대하여 반경험적 양자역학의 연산을 적용할 수 있는 것이다.
플라본과 6종의 플라본 유도체(Fig. 1)에 대하여 다음 단계에 서는 반경험적 모델 중 2주기 내의 원소들로 구성된 유기분자에
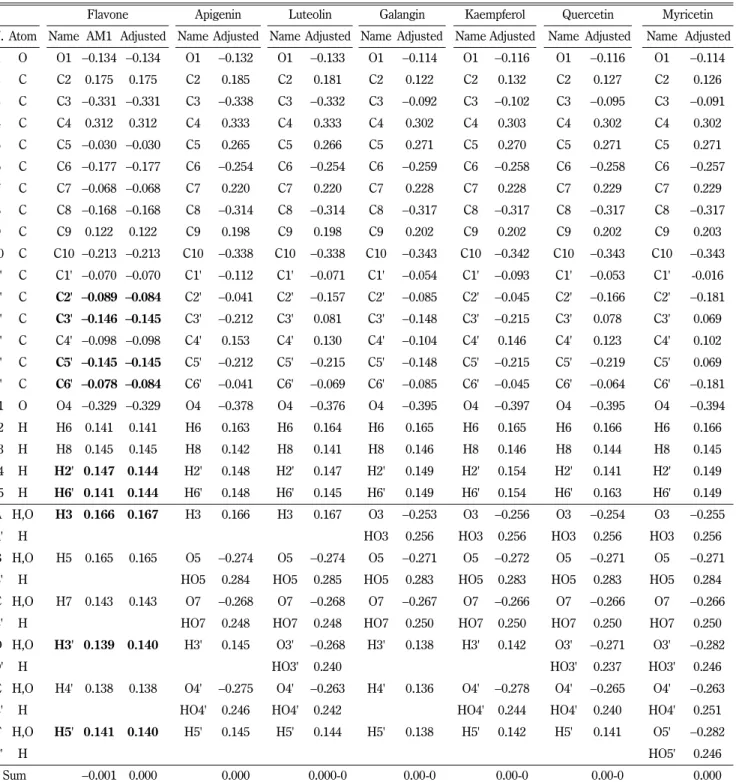
Table I − The atomic charges of flavone and flavone derivatives calculated with AM1 model
Flavone Apigenin Luteolin Galangin Kaempferol Quercetin Myricetin
N. Atom Name AM1 Adjusted Name Adjusted Name Adjusted Name Adjusted Name Adjusted Name Adjusted Name Adjusted 1 O O1 −0.134 −0.134 O1 −0.132 O1 −0.133 O1 −0.114 O1 −0.116 O1 −0.116 O1 −0.114
2 C C2 0.175 0.175 C2 0.185 C2 0.181 C2 0.122 C2 0.132 C2 0.127 C2 0.126
3 C C3 −0.331 −0.331 C3 −0.338 C3 −0.332 C3 −0.092 C3 −0.102 C3 −0.095 C3 −0.091
4 C C4 0.312 0.312 C4 0.333 C4 0.333 C4 0.302 C4 0.303 C4 0.302 C4 0.302
5 C C5 −0.030 −0.030 C5 0.265 C5 0.266 C5 0.271 C5 0.270 C5 0.271 C5 0.271
6 C C6 −0.177 −0.177 C6 −0.254 C6 −0.254 C6 −0.259 C6 −0.258 C6 −0.258 C6 −0.257
7 C C7 −0.068 −0.068 C7 0.220 C7 0.220 C7 0.228 C7 0.228 C7 0.229 C7 0.229
8 C C8 −0.168 −0.168 C8 −0.314 C8 −0.314 C8 −0.317 C8 −0.317 C8 −0.317 C8 −0.317
9 C C9 0.122 0.122 C9 0.198 C9 0.198 C9 0.202 C9 0.202 C9 0.202 C9 0.203
10 C C10 −0.213 −0.213 C10 −0.338 C10 −0.338 C10 −0.343 C10 −0.342 C10 −0.343 C10 −0.343 1' C C1' −0.070 −0.070 C1' −0.112 C1' −0.071 C1' −0.054 C1' −0.093 C1' −0.053 C1' -0.016 2' C C2' −0.089 −0.084 C2' −0.041 C2' −0.157 C2' −0.085 C2' −0.045 C2' −0.166 C2' −0.181 3' C C3' −0.146 −0.145 C3' −0.212 C3' 0.081 C3' −0.148 C3' −0.215 C3' 0.078 C3' 0.069 4' C C4' −0.098 −0.098 C4' 0.153 C4' 0.130 C4' −0.104 C4' 0.146 C4' 0.123 C4' 0.102 5' C C5' −0.145 −0.145 C5' −0.212 C5' −0.215 C5' −0.148 C5' −0.215 C5' −0.219 C5' 0.069 6' C C6' −0.078 −0.084 C6' −0.041 C6' −0.069 C6' −0.085 C6' −0.045 C6' −0.064 C6' −0.181 11 O O4 −0.329 −0.329 O4 −0.378 O4 −0.376 O4 −0.395 O4 −0.397 O4 −0.395 O4 −0.394
12 H H6 0.141 0.141 H6 0.163 H6 0.164 H6 0.165 H6 0.165 H6 0.166 H6 0.166
13 H H8 0.145 0.145 H8 0.142 H8 0.141 H8 0.146 H8 0.146 H8 0.144 H8 0.145
14 H H2' 0.147 0.144 H2' 0.148 H2' 0.147 H2' 0.149 H2' 0.154 H2' 0.141 H2' 0.149 15 H H6' 0.141 0.144 H6' 0.148 H6' 0.145 H6' 0.149 H6' 0.154 H6' 0.163 H6' 0.149 A H,O H3 0.166 0.167 H3 0.166 H3 0.167 O3 −0.253 O3 −0.256 O3 −0.254 O3 −0.255
A' H HO3 0.256 HO3 0.256 HO3 0.256 HO3 0.256
B H,O H5 0.165 0.165 O5 −0.274 O5 −0.274 O5 −0.271 O5 −0.272 O5 −0.271 O5 −0.271
B' H HO5 0.284 HO5 0.285 HO5 0.283 HO5 0.283 HO5 0.283 HO5 0.284
C H,O H7 0.143 0.143 O7 −0.268 O7 −0.268 O7 −0.267 O7 −0.266 O7 −0.266 O7 −0.266
C' H HO7 0.248 HO7 0.248 HO7 0.250 HO7 0.250 HO7 0.250 HO7 0.250
D H,O H3' 0.139 0.140 H3' 0.145 O3' −0.268 H3' 0.138 H3' 0.142 O3' −0.271 O3' −0.282
D' H HO3' 0.240 HO3' 0.237 HO3' 0.246
E H,O H4' 0.138 0.138 O4' −0.275 O4' −0.263 H4' 0.136 O4' −0.278 O4' −0.265 O4' −0.263
E' H HO4' 0.246 HO4' 0.242 HO4' 0.244 HO4' 0.240 HO4' 0.251
F H,O H5' 0.141 0.140 H5' 0.145 H5' 0.144 H5' 0.138 H5' 0.142 H5' 0.141 O5' −0.282
F' H HO5' 0.246
Sum −0.001 0.000 0.000 0.000-0 0.00-0 0.00-0 0.00-0 0.000
최적화된 오스틴모델 (AM1)을 적용하여 단일 단계 연산을 수행 하였다. 이 계산과정은 본 연구진의 지난 논문에 자세히 언급되 어 있고7)또한 Merck Index 나 NCI 데이터베이스에 저장된 모 든 화합물 군에도 적용되어 검증받은 방법이며8) 그 결과는 Table I에 수록하였다.
그 연산과정의 올바름을 검토하기 위하여 중성분자 전체의 총 전하의 값을 검토한 결과 일부 구조에서는 정확한 0의 값 대신 − 0.002, −0.001 또는 +0.001 등의 값을 보였는데 이 작은 편차는 어떠한 일관된 근거를 추정할 수 없을 정도로 산발적으로 관찰되 며 이는 양자역학적 연산과정 중에 간혹 관찰되는 연산오차(artifact) 로 추정되었으며 이를 보정하기 위하여 분자 내 구성 원소들의 대칭성을 검토한 후, 동등한 화학적 환경을 가진 원자의 전하를 동일하게 수정하므로써 총전하가 0이 되도록 조정(adjust)하였다.
이 조정과정은 Table I의 Flavone 분자의 경우를 모델로 AM1 계 산값과 조정값 (adjusted)을 굵은 글자체로서 대비하여 놓았다.
결과 및 고찰 (Results and Discussion)
이 연구에 포함된 플라본 유도체들의 전체적 계산 결과는 일 반적으로 알려진 원자의 전기음성도의 상대적 크기에 합당한 결 과를 보였다(Table I). 여기서 2'과 6', 3'와 5', 14'와 15', D와 F
는 입체적으로 동일한 위치와 화학적 환경에 처해 있으므로 치 환기가 있을 경우 연산오류를 제거하기 위해 각각 원자전하의 평 균을 구했고 전체 전하가 0으로 합산되도록 보정하여 최종값을 구하였다. 그 하나로 플라본에서 D(H3')와 F(H5')는 각각 0.139, 0.141의 계산값이 나왔지만 입체적으로 동일하므로 평균값인 0.140으로 보정하였다. 전체적으로 원자에 따른 전하를 살펴보면 산소는 음의 전하(−0.114 ~ −0.395), 탄소는 상황에 따라서 양 또는 음의 전하, 수소는 양의 전하를 띄고 있는데 이는 전기음성 도 순서와 일치한다. 이중 산소의 전하는 카보닐기로서 탄소와 이중결합을 이룰 때 −0.329 ~ −0.395, 수산기 같은 단일결합인 경우에는 −0.254 ~ −0.297, 에테르 결합을 할 때에는 −0.114 ~
−0.134 순으로 전하량이 감소하였음을 볼 수 있다.
1번 위치의 산소로 인하여 때문에 모든 유도체에서 2 (C2)와 9 (C9)위치의 탄소가 양으로 하전되었음을 확인할 수 있다. C2 탄소는 +0.122 ~ 0.185의 전하를 갖고 C9 탄소는 플라본을 제 외한 나머지 유도체에서 +0.198 ~ 0.203의 전하로 변화량이 적 었다. 이는 9 위치의 C9 탄소가 벤젠 고리를 구성하고 있는데 벤젠고리의 안정성으로 전하가 분산된 현상으로 볼 수 있다. 유 사한 이유로 5의 C5 수산기가 치환되어도 C10의 탄소는 전하가
−0.338 ~ 0.342로 유사한 전하값을 보임을 확인할 수 있다.
전체적으로 수산기가 치환됨으로써 인접하는 탄소의 순서대로
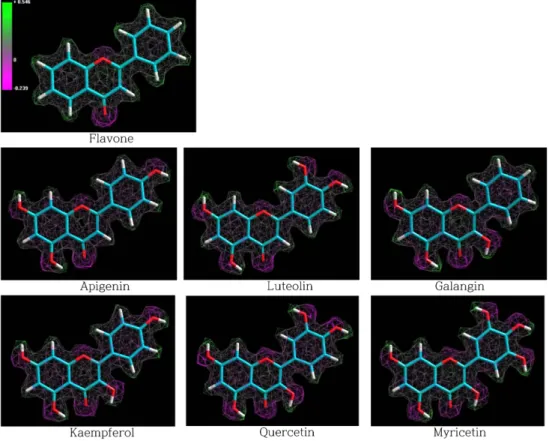
Fig. 2 − Atomic charge distribution of the flavone derivatives: the positive (green) and negative (violet) charge values is represented as 9000
grid points and connected lines with the grid distance of 0.5 Å.
각각 전하의 변화 부호가 +, −, +, −임을 확인할 수 있으며 플 라본을 제외한 다른 유도체에서 6의 C6가 벤젠 링에 있음에도
−0.254 ~ −0.259 전하로 다른 탄소보다 강하게 하전 되었는데 5, 7위치의 수산기에 의한 이중 효과의 결과임을 알 수 있다. C10 탄소도 C4의 카보닐기, 5의 수산기의 이중 효과로 −0.338 ~ −0.343 로 강한 음전하를 보여주는데 이번에는 카보닐기의 강한 극성 때문 에 더 강하게 하전되어 있는 양상이다.
전하계산은 반경험적 방법이나 ab initio 양자역학적 방법 등 여러 방법이 있으며 적용 방법에 따라 결과는 다를 수 있다. 실 제 Gaussian 프로그램을 사용한 계산에서는 위의 결과에 비하여 2배 이상의 강한 전하분포를 보였다. 이를테면 아미노산 타이로 신의 원자전하를 Gaussian 프로그램의 ab initio 법으로 계산해 보면 페놀기의 OH기의 O 및 H원자 대하여 각각 −2.120 및 +0.991의 극단적인 전하의 차이를 나타내고 반경험적 오스틴 모 델 (AM1)에 의하면 각각 −0.260 및 +0.221의 값을 보이는 것 을9)비교하면 전자간의 상호작용을 구체적으로 계산에 포함시키 는 ab initio 법에서는 확연히 전하의 비중을 과장하여 고려함을 확인할 수 있다. 타이로신과 유사한 페놀기를 가진 Apigenin의 전하값은 위 Table I에 O4‘ (−0.275) 및 HO4’ (+0.246)로서 유 사한 경항을 나타내고 있는 것이다.
이는 기존의 아미노산 분자에 대하여 적용하여도 유사한 결과 를 얻게 되는데 일반적으로 분자역학적 모델링에서 사용하는 힘 장에서는 강한 전하는 사용되지 않는다. 그 근거는 분자 간 상호 작용이 지나치게 과장되어 해석되는 경향이 있기 때문이며 AMBER10), CHARMM11)및 OPLS12)힘장 등 거대 생체분자를 모델링하는 프로그램에서는 완화된 전하, 즉, 반경험적 계산의 결과를 사용하는 것이다.
이 연구에서 계산된 전하들은 AMBER의 토폴로지 데이터에 저장된 아미노산들의 전하와 균형이 맞는다는 사실을 확인한 결 과이다. 즉, 플라본과 나프탈렌 분자 내의 원자와 형태가 같은 원 자들의 전하는 같은 결과를 보였다는 것이며 단백질과 같이 거 대한 생체분자를 포함하는 분자역학적 모델링에서는 AM1에 의 한 전하의 계산이 합리적인 방법임을 확인할 수 있었다.
계산된 전하분포의 3차원적 형상을 Fig. 2에 이소표면 (isosurface)의 형태로 제시하였다. 평균 9000개의 격자점을 원자 반경의 한도 안에서 0.5 Å의 간격으로 표시하여 양전하(초록생) 와 음전하(보라색)의 분포를 나타내었다. 플라본 유도체의 전체 구조는 거의 평면을 이루지만 OH 기가 도입된 부분에서는 O-C 결합주위의 이면각은 자유로이 회전 가능하므로 OH 말단 부분 의 전하분포는 가변성을 내포하고 있다.
결 론 (Conclusion)
유기분자의 물리적 성질 중 구성원자의 극성, 즉, 원자의 부분
전하로 표현되는 전기적 특성은 분자의 총체적 특성을 결정짓는 중요 요소 중의 하나이다. 이 특성은 용해도와 같은 분자 자체의 주변과의 관계에서 뿐 아니라 생리활성을 가지는 유기물 분자의 경우 그 활성의 수용체와의 관계에서는 작용의 본질적인 기능과 관계된 것이기도 하다. 이와 같은 수용체 단백질과 리간드의 상 호작용을 예측하고 계산하는 모의실험적 도구로는 일반적으로 고전역학의 분자역학적 알고리즘을 사용하는데, 이 경우 분자 내 원자의 부분전하와 같이 전자의 행동과 관련된 특성은 고전역학 으로는 자체 계산이 불가능하므로 양자역학적으로 계산한 결과 를 전환하여 반영해야 하는 것이다. 본 연구에서 수행한 AM1 모 델에 의한 반경험적 방법으로 계산한 원자들의 전하는 기존의 분 자역학적 모델인 AMBER나 CHARMM에서 사용되는 주요 생 체분자들의 원자전하와 일관적인 강도의 원자전하를 산출하는 것으로 파악되었으며 앞으로도 임의의 유기분자들의 합리적인 원자 전하는 이와 같은 경로를 통하여 산출하는 것이 좋을 것으 로 판단된다.
이 연구의 결과는 향후 앞으로 서론에서 언급한 플라본류의 생리활성물질이 분자표적과의 상호작용에서 어떤 역할을 하는가 를 연구하는 중간 단계의 과정에 중요한 수단으로 활용될 것으 로 기대된다.
감사의 말씀 (Acknowledgment)
이 연구는 충남대학교의 학술연구비 지원에 의한 것으로 이에 감사를 표한다.
References
1) Malesev, D. and Kuntic, V. : Investigation of metal-flavonoid chelates and the determination of flavonoids via metal- flavonoid complexing reactions. J. Serbian Chem. Soc. 72, 922 (2007).
2) Rice-Evans, C. : Flavonoid antioxidants. Current Med. Chem..
8, 797 (2001).
3) Pietta, P. G. : Flavonoids as antioxidants. J. Nat. Prod. 63, 1035 (2000).
4) Stewart, J. J. P. MOPAC : A semiempirical molecular orbital program. J. Comput. Aided Mol. Des. 4, 1 (1990).
5) Hyperchem (TM) Professional 8.0, Hypercube, Inc., NW 4th Street, Gainesville, Florida 32601, USA (2010).
6) Allinger, N. L. : Molecular mechanics force field. J. Am. Chem.
Soc. 99, 8127 (1977).
7) Park, K. L. : Modeling partial atomic charge of organic molecule and mutated amino acid in a protein-ligand complex for molecular mechanics simulation. Bull. Korean Chem. Soc.
34, 299 (2013).
8) Jakalian, A., Bush, B. L., Jack, D. B. and Bayly, C. I. : Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 21, 132 (2000).
9) Park, K. L. : Unpublished work. (2012).
10) Case, D. A., Darden, T. A., Cheatham III, T. E., Simmerling, C.
L., Wang, J., Duke, R. E., Luo, R., Walker, R. C., Zhange, W., Merz, K. M., Roberts, B., Wang, B., Hayik, S., Roitberg, A, Seabra, G., Kollosvary, I., Wong, K. F., Paesani, F., Vanicek, J., Liu, J., Wu, X., Brozell, S. R., Steinbrecher, T.,Goehlke , H., Cai, Q., Ye, X., Wang, J., Hsieh, M. J., Cui, G., Roe, D. R., Mathews, D. H., Seetin, M. G., Sagui, C., Babin, V., Luchko, T.,
Gusarov, S., Kovalenko, A. and Kollman, P. A. : AMBER12, University of California, San Francisco (2012).
11) Brooks, B. R., Bruccoleri, R. E., Olasfson, B. D., States, D. J., Swaminathan, S. and Karplus, M. : CHARMM: A program for macromolecular energy, minimization, and dynamics calculations J. Comput. Chem. 4, 187 (1983).
12) Jorgensen, W. L, Maxwell, D. S. and Tirado-Rives, J. : Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J.
Am. Chem. Soc. 118, 11225 (1996).