I. INTRODUCTION
A. Bcl-xL-mediated ERK2 activation plays a critical role in the inhibition of
doxorubicin-induced apoptosis
Bcl-xL overexpressed in various tumors confers resistance to various chemotherapeutic agents (Zapata JM et al., 2003, McConkey DJ et al., 1996; Tang L
et al., 1998; Takehara T et al., 2001). Anti-apoptotic activity of Bcl-xL has been
reported to be closely associated with the prevention of the release of mitochondrial cytochrome c into cytosol following exposure to anti-cancer drugs (Kirkin V et al., 2004). However, it is not well characterized whether resistance of Bcl-xL-overexpressing cancer cells to chemotherapeutic agents is associated with the modulation of the activities of signaling molecules. Doxorubicin, anthracyclin antibiotics, is one of the most effective chemotherapeutic agents against a wide variety of cancers (Hortobagyi GN, 1997). Doxorubicin has been reported to act through DNA intercalation/binding, inhibition of topoisomerase II, free radical generation, or damage to cell membranes (Gewirtz DA, 1999). However, anti-proliferative and death-inducing mechanisms of doxorubicin are still far from well characterized, despite its widespread clinical use.
MAP kinase signaling pathways modulate gene expression, mitosis, proliferation, motility, metabolism and apoptosis (Wada T and Penninger JM, 2004). MAP kinase consists of three family members: the extracellular signal-regulated kinase (ERK);
the c-Jun NH2-terminal kinase (JNK); and the p38. The potential involvement of JNK and p38 pathways in both proliferative and apoptotic parhways make prediction of the involvement of JNK and p38 signaling in cancer more complex. (Wada T and Penninger JM, 2004). However, ERK pathways are well known to be involved in tumor cell survival and/or proliferation and thus may be potential targets for cancer therapy (Chang F et al., 2003). In this study, we investigated whether anti-apoptotic effect of Bcl-xL on doxorubicin-induced apoptosis is linked to the control of MAP kinase signaling pathways.
ERK2 was significantly activated in various Bcl-xL-overexpressing cells treated with doxorbicin, whereas doxorubicin-induced activation of JNK and p38 was not affected by Bcl-xL overexpression. In this study, we for the first time presented evidences that ERK2 activation mediated by Bcl-xL plays a critical role in blocking of doxorubicin-induced apoptosis.
B. Bcl-xL blocks apoptosis but not mitotic cell death induced by doxorubicin
Apoptotic defects play roles in multiple ways to tumor progression (Reed JC, 1999) and tumor resistance to chemotherapy (Kitada S et al., 2002). Thus, apoptosis has been considered as a major target of cancer therapy. However, apoptosis seems to have little or no influence on chemotherapy outcome for other solid tumors (Brown JM and Wouters BG, 1999; Roninson IB, 2001).
Mitotic cell death is the form of cell death that results from aberrant mitosis (Roninson IB, 2001). This cell death is associated with formation of nonviable cells
with several micronuclei that are formed from nuclear membrane encapsulation of chromatin and chromosomal fragments that fail to properly segregate after mitosis (Müller WU et al., 1996). Mitotic cell death has been characterized as the main form of cell death induced by ionizing radiation (Jonathan EC et al., 1999), and is identified as a prominent response to different anticancer drugs, such as etoposide, taxol, cisplatin or bleomycin (Tounekti O et al., 1993).
Hepatocellular carcinoma (HCC) is one of the most common cancers and many of them cause cancer-related death (Parkin DM et al., 2001).Recently, the incidence of HCC is increasing world-wide and HCC is the third most common cause of cancer-related death, therefore advances in prevention and treatment of HCC are a major health priority (Pons-Renedo F and Llovet JM, 2003). HCC is resistant to various apoptotic stimuli. Bcl-xL, an antiapoptotic member of Bcl-2 family, is highly expressed in human HCC. High expression of endogenous Bcl-xL in HCC-derived cell lines is important for the inhibition of apoptosis that is initiated by various cellular stresses (Takehara T et al., 2001). It has not been clarified whether Bcl-xL affects mitotic cell death, while the blocking effect of Bcl-xL on apoptosis is well defined.
Our previous report suggests that high doses of doxorubicin, anthracycline antibiotics which acts through DNA intercalation/binding, inhibition of topoisomerase II, free radical generation, or damage to cell membranes (Gewirtz DA, 1999), induce apoptosis while low doses of doxorubicin induce mitotic cell death accompanying senescence-like phenotype (SLP).
Here, we investigated the effect of Bcl-xL on LD doxorubicin-induced SLP and mitotic cell death in HCC cells. We found that induction of SLP and mitotic cell death by LD doxorubicin is slightly delayed but not blocked by Bcl-xL overexpression. Therefore, induction of SLP and/or mitotic cell death by LD doxorubicin may be an alternative strategy for Bcl-xL-overexpressing HCC cells, which are resistant to apoptotic effect of chemotherapeutic agents.
III. MATHERIALS AND METHODS
A. Chemicals and antibiotics
Doxorubicin, PD98059, U0126 were from Sigma (Sigma, St. Louis, MO). Calcein-AM, Etd-1, 4',6-diamidino-2-phenylindole, dihydrochloride (DAPI), propidium iodide (PI), 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethyl-benzamidazolocarbocyanin iodide (JC-1), Rhodamine 123, fluorescein isothiocynate (FITC)-conjugated phalloidin and Texas-Red-conjugated phalloidin were from Molecular probe (Eugen, OR). The following antibodies were used: anti-α-tubulin, caspase-9, Lamin B, Cdc2, Cdc25c, Cyclin A and Cyclin B (Santacruz Biotechnologies, Santacruz, CA); anti-MAD2, CENP-A, phospho-p38, total p38, phospho-JNK, total JNK, phospho-ERK, total ERK, phospho-p90RSK, phospho-CREB, phospho-p65 and phospho-MEK (Cell
signaling, Beverly, MA); anti-caspase-8, caspase-3 and Smac (Stressgen, British Columbia, Canada); anti-cytochrome c (BD Pharmingen, San Diego, CA); anti-Flag M2, FITC-conjugated anti-mouse IgG and FITC-conjugated anti-goat IgG, (Sigma, St Louis, MO); anti-rabbit IgG HRP, mouse IgG HRP and goat IgG HRP(Zymed Laboratories, Inc., South San Francisco, CA)
B. Cell culture
Huh-7, Chang and U87MG cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO-BRL, Life Technologies, NY) supplemented with 10%
fetal bovine serum and antibiotics (GIBCO-BRL, Life Technologies, NY).
C. Measurement of cellular viability
Cell viability was assessed by double labeling of cells with 2 µM calcein-AM and 4 µM Etd-1. The calcein-positive live cells and Etd-1-positive dead cells were visualized using a fluorescence microscope (Nikon Diaphot 300, Japan).
D. Bcl-xL-, Bcl-2-, XIAP-, Akt- or survivin overexpressing stable cell lines.
Mammalian expression vector containing Flag-tagged bcl-xL and bcl-2 (Huang DA et al., 1997) were kindly provided by A Strasser (The Walter and Eliza Hall Institute of Medical Research, Australia). Stable cell lines overexpressing Bcl-xL was selected with changes of fresh media containing puromycin (4 µg/ml) in Huh-7 and Chang cells. Overexpression of Bcl-xL in the stable cell lines was analyzed by western blotting using anti-Flag antibody. Establishment of stable cell lines overexpressing Bcl-2 in Huh-7 cells was performed as described in Bcl-xL overexpression. To compare the effect of Bcl-xL, Bcl-2, XIAP, Akt, survivin on doxorubicin-induced apoptosis, U87MG cells stably transfected with Bcl-xL, Bcl-2, XIAP, survivin (Kim EH et al., 2004b), or Akt (Kim EH et al., 2004a) were used.
E. Immunublotting
Cells were washed in PBS and lysed in boiling sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (62.5 mmol/L
Tris [pH 6.8], 1% SDS, 10% glycerol, and 5% β-mercaptoethanol). The lysates were boiled for 5 min, separated by SDS-PAGE, and transferred to an Immobilon membrane (Millipore, Bedford, MA). After blocking nonspecific binding sites for 1h with 5% skim milk, membranes were incubated for 2 h with specific Abs. Membranes were then washed three times with TBST and incubated further for 1h with horseradish peroxidase-conjugated anti rabbit, mouse or goat antibody. Visualization of protein bands was accomplished using ECL (Amersham Life Science, Buckinghamshire, UK).
F. Immunocytochemistry
All tested cells were seeded onto coverslips and incubated for 1 day before doxorubicin treatment. Cell were treated with doxorubicin for the indicated concentrations and times, and then fixed with 4% paraformaldehyde (pH 7.4) for 10 min at 4℃, permeabilized in 0.1% Triton X-100 in PBS for 5 min. Permeabilized cells were incubated for 45 min at room temperature with anti-Lamin B, cytochrome c, Smac antibodies, washed with phosphate buffered saline (PBS) 3 times, and then incubated with FITC-conjugated goat or mouse antibodies for 30 min at room temperature. Nuclei were further stained for 10 min in 1 µg/ml of DAPI or PI in PBS and actin was stained with FITC or Texas Red-conjugated phalloidin. Cells were subsequently mounted in fluorescent mounting medium (DAKO corp., CA) and visualized by confocal laser microscope.
G. SA-ββββ-gal activity
Following the experimental protocol by Dimri et al. (Dimri GP et al., 1995), the cells seeded on 24-well plate were treated with LD doxorubicin for different time points, washed twice with PBS, fixed with 2% formaldehyde and 0.2% glutaraldehyde in PBS, and washed twice in PBS. Cells were stained for 12 h in X-gal staining solution [1 mg/ml X-gal, 40 mmol/l citric acid/sodium phosphate (pH 6.0), 5 mmol/l potassium ferricyanide, 5mmol/L potassium ferrocyanide, 150 mmol/l NaCl, 2 mmol/l MgCl2]. Cells were then counterstained with 0.1% haematoxylin solution.
H. Detection of mitochondrial permeability transition
Changes in the mitochondrial membrane potential were detected using two fluorescent cationic dye, Rhodamine 123 and 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethyl-benzamidazolocarbocyanin iodide, commonly known as JC-1 (Molecular Probes, Eugene, OR). Rhodamine 123 is readily sequestered by functioning mitochondria and subsequently washed out of the cells once the mitochondrial membrane potential is lost (Nadakavukaren KK et al., 1985). Tested cells were seeded onto coverslips and treated with doxorubicin at the indicated concentrations and times. Treated cells were incubated with 1 µg/ml Rhodamine 123 in media for 30 min at 37℃. Cells were visualized by fluorescent microscope. JC-1 has dual
fluorescents. Inside healthy cells, the lipophilic positively charge JC-1 dye aggregates in mitochondria and under fluorescent light emits a re signal (J-aggregates emit at 590 nm) (Coluccia AM, 2004). Upon reduction in MMP, the JC-1fluorochrome converts to a monomeric form that fluoresces green (monomeric dye emits at 490nm). Cells were incubated with 2 µg/ml of JC-1 in media for 30 min at 37℃. All stained samples were analyzed using FACS.
I. Detection of ATP depletion
ATP was measured with an ATP Determination Kit (Molecular Probes, Eugene, OR), following the instructions of the manufacturer. In Brief, cells treated with doxorubicin at indicated concentrations and times were lysed with 1X Passive Lysis Buffer (Promega, Obispo, CA), and lysates were diluted 1:10 by using the ATP Determination Kit reaction mix for a total volume of 200 µl. Luminescence was determined directly after the addition of lysates to the reaction mixture and was quantified in a 20/20 Luminometer (Turner Designs, Sunnyvale, CA). ATP concentrations in the experimental samples were calculated by using an ATP standard curve.
J. Subcellular Fractionation
subcellular fractionation was performed (Akao Y et al., 1994). Harvested cells were washed twice with ice-cold PBS and then resuspended in hypotonic buffer (10 mmol/l HEPES, 10 mmol/l MgCl2, and 42 mmol/l KCl). Cells were passed through a
25-gauge syringe and centrifuged at 1,000 rpm for 5 minutes to remove unlysed cells and nuclei. The supernatant was further centrifuged at 15,000 rpm for 15 minutes at 4℃. The supernatant was ultracentrifuged at 100,000g for 15 minutes at 4℃. The supernatant was used as the cytosolic fraction. The respective fractions were processed for western blotting of cytochrome c or Smac.
K. Cell cycle analysis
DNA content was assessed using Cell Cycle TEST PLUS DNATM reagent kit
(Becton Dickinson and Co., San José, CA) according to the manufacturer’s instruction and analyzed in a FACS sorter (Becton Dickinson and Co.). Trypsinized and floating cells were pooled, washed with PBS-EDTA, and fixed in 70% (v/v) ethanol. DNA content was assessed by staining cells with propidium iodide and monitoring by FACS. Cell distribution was determined with a ModFit LT program (Verity Software House, Inc.)
L. Clonogenic assay
treatment with 50 ng/ml doxorubicin for the indicated time points. Culture media were replaced with fresh media not containing doxorubicin and cells were further incubated by 12 days. Clonogenic survival was determined by staining colonies using 0.5% crystal violet in 20% methanol for 20 min at room temperature and visualized by digital camera.
M. Expression of dominant-negative ERK2 or wild type ERK2 by transient
transfection
Chang cells or Bcl-xL-overexpressing Chang cells were plated into 24-well plates at (3 X 104) cells/well. After 24 h, cells were transfected with the expression vector
encoding dominant-negative ERK2 mutant at the indicated concentrations using Lipofectamine Plus reagent (Gibco BRL, Grand Island, NY) following the manufacturer’s instructions, Bcl-xL-overexpressing Chang cells were transfected with the expression vector encoding wild type ERK2. After incubation of the transfected cells for 24 h, cells were further treated with or not treated with 1 µg/ml of doxorubicin for 24 h. Transfection efficiency, which was assessed using the plasmid encoding green fluorescence protein, was up to 80% in Chang and Bcl-xL overexpressing Chang cells. Cellular viability was assessed using calcein-AM and Etd-1 as described above.
III. RESULTS
A. Bcl-xL-mediated ERK2 activation plays a critical role in the
inhibition of doxorubicin-induced apoptosis
1. Bcl-xL overexpression blocks doxorubicin-induced apoptosis in Huh-7
hepatoma cells
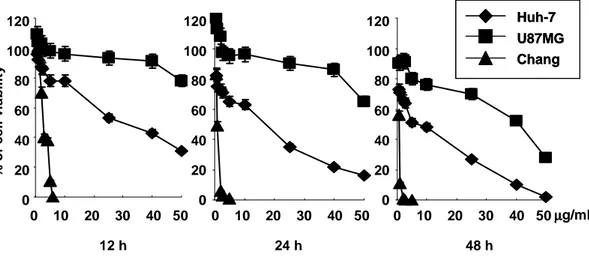
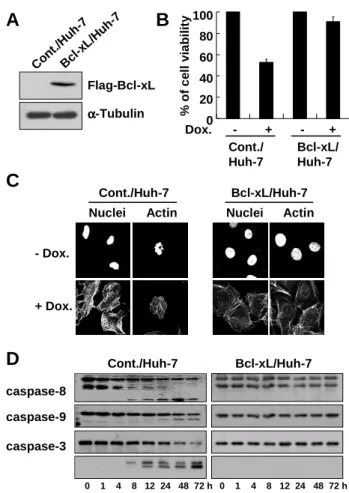
First, we investigated the effect of Bcl-xL overexpression on doxorubicin-induced apoptosis. While treatment with 10 µg/ml doxorubicin for 24 h resulted in the decrease of cellular viability by 50% in control Huh-7 cells (Fig. 1, Table 1), this cell killing effect of doxorubicin was significantly blocked in Huh-7 cells overexpressing Bcl-xL (Fig. 2B). Doxorubicin-induced reduction of cellular size, DNA fragmentation (Fig. 2C), and activation of caspases (Fig. 2D) were also completely blocked by Bcl-xL overexpression.
2. ERK2 is activated in Bcl-xL overexpressing Huh-7 cells treated with HD
doxorubicin
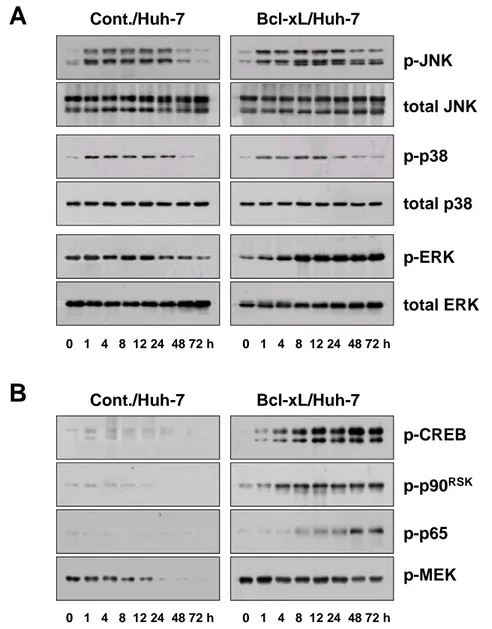
Next, we investigated whether blocking of doxorubicin-induced apoptosis by Bcl-xL overexpression was associated with the modulation of MAP kinase signaling pathways. Western blotting using specific antibodies for active JNK and p38 demonstrated that both JNK and p38 were rapidly activated by treatment with 10
µg/ml doxorubicin and their high activities were sustained by 24 h (Fig. 3A). Bcl-xL overexpression did not affect doxorubicin-induced activation of JNK and p38. However, ERK1/2 activities, in particular ERK2 activity, were significantly increased from 1 h of doxorubicin treatment and their high activities were maintained in Bcl-xL-overexpressing cells by 72 h, while ERK1/2 activities were gradually decreased in control Huh-7 cells treated with doxorubicin. CREB and p90RSK, the
downstream targets of ERK signaling pathway, were also significantly activated in Bcl-xL-overexpressing cells with the similar activation profiles of ERK, whereas their activities were downregulated in control Huh-7 cells treated with doxorubicin (Fig. 3B). Since ERK pathway has been recently implicated in doxorubicin-induced κB activation in (10)1 cells (Panta GR et al., 2004), we investigated whether NF-κB is activated in Bcl-xL-overexprssing cells treated with doxorubicin. Western blotting using specific antibody for active p65 demonstrated that protein levels of active p65 increased in Bcl-xL-overxressing cells from 8 h of doxorubicin treatment and its high activity was sustained by 72 h. However, the activities of MEK1/2, which are known as upstream activator of ERK, were not increased in parallel with ERK2 activity in Bcl-xL-overexpressing Huh-7 cells treated with doxorubicin, while they were decreased in control Huh-7 cells. These results suggest that Bcl-xL-mediated ERK2 activation in response to doxorubicin is independent of MEK1/2 activities.
apoptosis accompanying the activation of ERK2
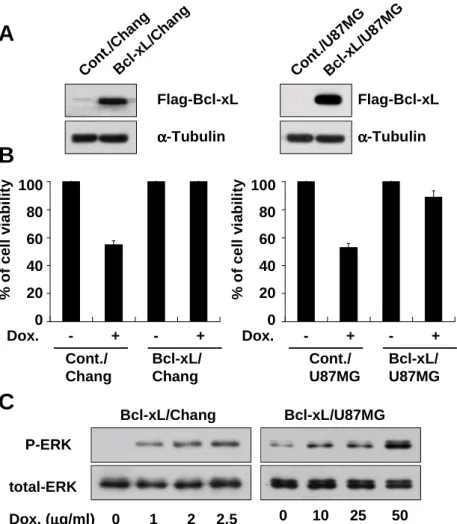
To analyze whether activation of ERK2 pathway in Bcl-xL overexpressing cells treatment with doxorubicin is restricted to a particular cell line, we examined the effect of Bcl-xL overexpression on doxorubicin-induced apoptosis and ERK2 activity in other cell lines. We first determined IC50, the concentration of doxorubicin
demonstrating 50% cytotoxicity after incubation for 24 h, in Chang liver cells and U87MG glioma cells and its value was 1 µg/ml and 40 µg/ml respectively (Fig. 1, Table. 1). We established the stable cell lines overexrpessing Bcl-xL in Chang cells and U87MG cells (Fig. 4A). Overexpression of Bcl-xL in both cell lines significantly blocked doxorubicin-induced apoptosis (Fig. 4B). Moreover, treatment with doxorubicin increased the activity of ERK, in particular ERK2, in a dose-dependent manner both in Bcl-xL-overexpressing Chang and U87MG cells (Fig. 4C). Taken together, our results demonstrated that ERK2 is commonly activated when doxorubicin-induced apoptosis is blocked by Bcl-xL overexpression.
120 100 80 60 40 20 0 120 100 80 60 40 20 0 120 100 80 60 40 20 0 % o f c e ll v ia b il it y 0 10 20 30 40 50 0 10 20 30 40 50 0 10 20 30 40 50µµµµg/ml Huh-7 U87MG Chang Huh-7 U87MG Chang 12 h 24 h 48 h
Fig. 1. Chemosensitivity of Huh-7, U87MG and Chang cells toward doxorubicin treatment. To evaluate the drug sensitivity, Live/Dead assay was performed in cells 12, 24 and 48 h after doxorubicin treatment respectively. The results are mean values of three independent experiments yielding similar results.
3.3 0.05 0.9 0.075 0.02 0.003 Normal liver Chang 58 4.5 36.3 0.300 0.4 0.100 Glioma U87MG 39.3 1.4 11 1.000 0.05 0.002 Hepatoma Huh-7 IC90(µµµµg/ml) IC50(µµµµg/ml) IC10(µµµµg/ml) Cell type Cell line 3.3 0.05 0.9 0.075 0.02 0.003 Normal liver Chang 58 4.5 36.3 0.300 0.4 0.100 Glioma U87MG 39.3 1.4 11 1.000 0.05 0.002 Hepatoma Huh-7 IC90(µµµµg/ml) IC50(µµµµg/ml) IC10(µµµµg/ml) Cell type Cell line
Table 1. Determination of the cytotoxic effects of doxorubicin on Huh-7, U87MG and Chang cells. IC10, IC50 and IC90 are the concentrations of doxorubicin giving
10%, 50% and 90% growth inhibition, respectively, in the Live/Dead assay at 24 h in Huh-7, U87MG cells and Chang cells after doxorubicin treatment. Values are SD mean in micrograms per milliliter from at least three independent experiments.
% o f c e ll v ia b il it y 100 80 60 40 20 0 Cont./ Huh-7 Bcl-xL/ Huh-7 A Flag-Bcl-xL α αα α-Tubulin B Dox. - + - + C Cont./Huh-7 Bcl-xL/Huh-7 - Dox. + Dox.
Nuclei Actin Nuclei Actin
D caspase-8 caspase-9 caspase-3 Cont./Huh-7 Bcl-xL/Huh-7 0 1 4 8 12 24 48 72 h Con t./H uh-7 Bcl -xL/ Huh -7 0 1 4 8 12 24 48 72 h
Fig. 2. Bcl-xL blocks doxorubicin-induced apoptosis. (A) Expression of Bcl-xL in the stable cell lines overexpressing Flag-tagged Bcl-xL. To detect the expression of stably transfected Bcl-xL, Western blotting using anti-Flag was performed. Expression of α-tubulin was monitored as a loading control. (B) Blocking of doxorubicin-induced cell death by xL overexpression. Cont./Huh-7 and Bcl-xL/Huh-7 cells were treated with 10 µg/ml doxorubicin and cellular viability was measured using calcein-AM and EthD-1. (C) Effect of Bcl-xL overexprssion on doxorubicin-induced activation of caspases. Cont./Huh-7 or Bcl-xL/Huh-7 cells were treated with 10 µg/ml of doxorubicin for 24 h were fixed and stained for nuclei and actin using PI and FITC-conjugated phalloidin, respectively. (D) Effect of Bcl-xL overexpression on doxorubicin-induced activation of caspases. Cont./Huh-7 or Bcl-xL/Huh-7 cells were treated with 10 µg/ml doxorubicin for the indicated time points and cell extracts were prepared for Western blotting of caspase-8, -9 and -3 was performed. Expression of CDK4 was monitored as a loading control.
p-JNK total JNK p-p38 total p38 Cont./Huh-7 Bcl-xL/Huh-7 p-ERK total ERK
A
B
p-CREB p-p90RSK p-MEK Cont./Huh-7 Bcl-xL/Huh-7 p-p65 0 1 4 8 12 24 48 72 h 0 1 4 8 12 24 48 72 h 0 1 4 8 12 24 48 72 h 0 1 4 8 12 24 48 72 hFig. 3. ERK2 is activated in Bcl-xL-overexpressing cells treated with doxorubicin. (A) Effect of Bcl-xL overexpression on the activities of MAP kinase following treatment with doxorubicin. Control Huh-7 cells and Bcl-xL/Huh-7 cells were treated with 10 µg/ml doxorubicin for the indicated time periods and cell extracts were prepared for Western blotting to detect the activities and total expression of MAP kinases. (b) Effect of Bcl-xL overexpression on the activities of the signaling molecules of ERK pathway after doxorubicin treatment. Changes in CREB, p90RSK, p65 and MEK1/2 activities were examined by Western blotting using the respective phospho-specific antibodies.
Dox. (µµµµg/ml) 0 1 2 2.5
A
Con t./C hang Bcl -xL/ Cha ng Flag-Bcl-xL α αα α-Tubulin Con t./U 87M G Bcl -xL/ U87 MGB
% o f c e ll v ia b il it y 100 80 60 40 20 0 Cont./ Chang Bcl-xL/ Chang Dox. - + - + % o f c e ll v ia b il it y 100 80 60 40 20 0 Cont./ U87MG Bcl-xL/ U87MG Dox. - + - +C
Bcl-xL/Chang Bcl-xL/U87MG 0 10 25 50 P-ERK total-ERK Flag-Bcl-xL α αα α-TubulinFig. 4. Bcl-xL overexpression in Chang or U87MG cells blocks doxorubicin-induced apoptosis accompanying the activation of ERK2. (A) Bcl-xL expression in the stable cell lines overexpressing Flag-tagged Bcl-xL. Western blotting using anti-Flag antibody was performed to detect the stably transfected Flag-tagged Bcl-xL. (B) Blocking of doxorubicin-induced cell death by Bcl-xL overexpression in Chang or U87MG cells. Control Chang cells or Bcl-xL/Chang cell were treated with 1 µg/ml doxorubicin for 24 h. Control U87MG cells or Bcl-xL/U87MG cells were treated with 40 µg/ml doxorubicin for 24 h and cellular viability was measured using Calcein-AM and EthD-1. (C) Doxorubicin-induced ERK2 activation in Bcl-xL-overexpressing Chang or U87MG cells. Bcl-xL/Chang and Bcl-xL/U87MG cells were treated with doxorubicin as the indicated concentration for 24 h and cell extracts were prepared for Western blotting to detect the activities and total expression of ERK.
4. ERK2 activation is important for the blocking effect of Bcl-xL on
doxorubicin-induced apoptosis
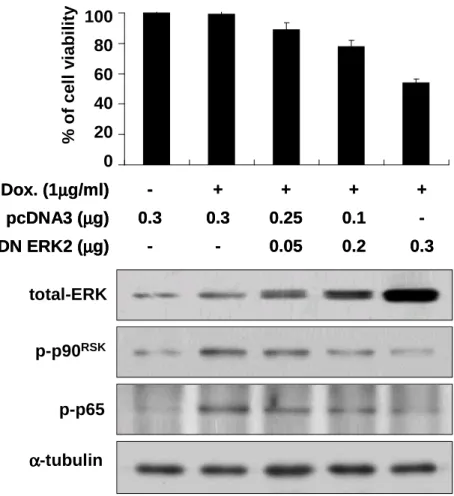
Next, we investigated the significance of ERK2 activation in Bcl-xL-overexpressing cells treated with doxorubicin. We first examined whether forced expression of a DN ERK2 mutant could attenuate the death-blocking effect of Bcl-xL following exposure to doxorubicin. Bcl-xL-overexpressing Chang cells were transiently transfected with pcDNA3 and/or the plasmid encoding DN erk2 mutant and further treated with doxorubicin (Fig. 5). Forced expression of DN ERK2 mutant decreased the activity of endogenous p90RSK in a dose dependent manner, when we assed p90RSK activity
by Western blotting using phospho-specific antibody of p90RSK. At the same time,
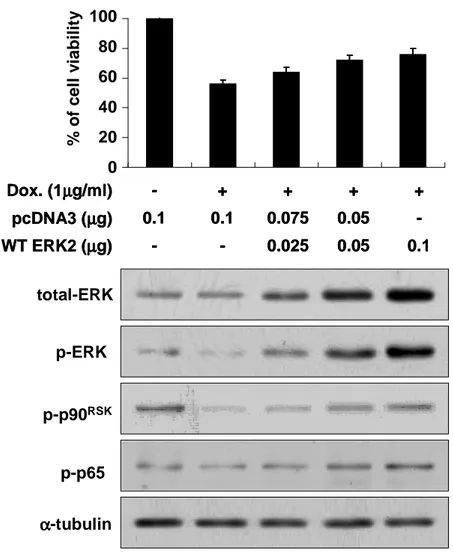
protein levels of active p65, which was measured by Western blotting using the specific antibody for active p65, were also downregulated by enhanced expression of DN ERK2 mutant. Inhibition of ERK2 activity by expression of DN ERK significantly reduced cellular viability in Bcl-xL-overexpressing cells treated with doxorubicin. Next, we further tested whether forced expression of WT ERK2 could attenuate doxorubicin-induced apoptosis in control Chang cells (Fig. 6). Transfection of Chang cells with the plasmid encoding WT ERK2 increased the activities of ERK2 and p90RSK and doxorubicin-induced cell death was significantly attenuated in
a dose-dependent manner of WT ERK2. Protein levels of active p65 were also enhanced by forced expression of WT ERK2. Taken together, ERK2 activated in Bcl-xL-overexpressing cells following exposure to doxorubicin plays a key role in the blocking of cell death. ERK2-mediated NF-κB activation may also contribute to
strong resistance to doxorubicin in Bcl-xL-overexpressing cells.
5. Activation of ERK2 can inhibit the release of mitochondrial cytochrome c
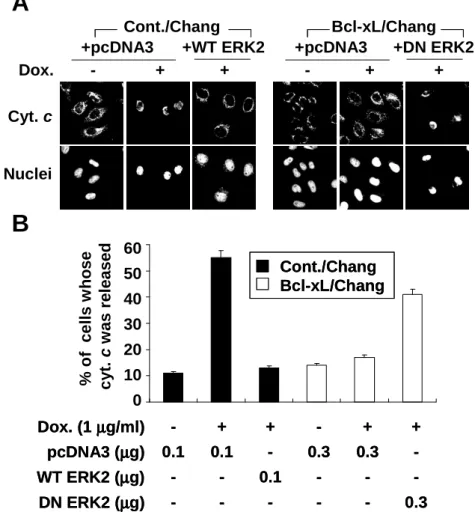
Next, we investigated whether ERK2 activated by doxorubicin in Bcl-xL-overexpressing cells affected the release of mitochondrial cytochrome c into cytosol. Immunocytochemistry using anti-cytochrome c antibody demonstrated that diffuse forms of cytochrome c were detected in control cells treated with doxorubicin, demonstrating the release of mitochondrial cytochrome c (Fig. 7A). However, punctuate forms of cytochrome c were detected in cells overexpressing Bcl-xL treated with doxorubicin, suggesting that release of mitochondrial cytochrome c was blocked by Bcl-xL overexpression. In Bcl-xL-overexpressing Chang cells transfected with DN ERK2, the percentages of cells whose cytochrome c was released from mitochondria were significantly increased by doxorubicin treatment, while transfection with DN ERK2 without doxorubicin treatment did not significantly affect it. Moreover, in control Chang cells transfected with WT ERK2, the percentages of cells whose cytochrome c was released from mitochondria were significantly decreased following doxorubicin treatment. These results suggest that ERK2 activated by doxorubicin in Bcl-xL-overexpressing ells may act upstream of the mitochondrial levels.
% o f c e ll v ia b il it y 100 80 60 40 20 0 α αα α-tubulin total-ERK p-p90RSK 0.3 0.2 0.05 -DN ERK2 (µµµµg) -0.1 0.25 0.3 0.3 pcDNA3 (µµµµg) + + + + -Dox. (1µµµµg/ml) 0.3 0.2 0.05 -DN ERK2 (µµµµg) -0.1 0.25 0.3 0.3 pcDNA3 (µµµµg) + + + + -Dox. (1µµµµg/ml) p-p65
Fig. 5. Forced expression of DN ERK2 enhances doxorubicin-induced apoptosis in Bcl-xL-overexpressing cells. Bcl-xL/Chang cells were trasfeceted with the plasmid encoding DN ERK2 and/or pcDNA3 at the indicated concentrations and further treated with or without 1 µg/ml doxorubicin for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. To confirm whether the transfected DN ERK2 was functioning, cell extracts prepared from the transfected cells were processed for Western blotting to detect total ERK protein levels, activities of p90 RSK and p65. Expression of α-tubulin was monitored as a loading control
0.1 0.05 0.025 -WT ERK2 (µµµµg) -0.05 0.075 0.1 0.1 pcDNA3 (µµµµg) + + + + -Dox. (1µµµµg/ml) 0.1 0.05 0.025 -WT ERK2 (µµµµg) -0.05 0.075 0.1 0.1 pcDNA3 (µµµµg) + + + + -Dox. (1µµµµg/ml) α αα α-tubulin total-ERK p-p90RSK p-ERK 100 80 60 40 20 0 p-p65 % o f c e ll v ia b il it y
Fig. 6. Forced expression of WT ERK2 attenuated doxorubicin-induced apoptosis in Chang cells. Chang cells were trasfeceted with the plasmid encoding DN ERK2 and/or pcDNA3 at the indicated concentrations and further treated with or without 1 µg/ml doxorubicin for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. To confirm whether the transfected DN ERK2 was functioning, cell extracts prepared from the transfected cells were processed for Western blotting to detect total ERK protein levels, activities of ERK, p90 RSK and p65. Expression of α-tubulin was monitored as a loading control
+pcDNA3 Dox. - + + +WT ERK2 Cyt. c Nuclei - + + +pcDNA3 +DN ERK2 Cont./Chang Bcl-xL/Chang % o f c e ll s w ho s e c y t. c w a s r e le a s e d 60 50 40 30 20 10 0 0.3 -DN ERK2 (µµµµg) -0.1 -WT ERK2 (µµµµg) -0.3 0.3 -0.1 0.1 pcDNA3 (µµµµg) + + -+ + -Dox. (1 µµµµg/ml) 0.3 -DN ERK2 (µµµµg) -0.1 -WT ERK2 (µµµµg) -0.3 0.3 -0.1 0.1 pcDNA3 (µµµµg) + + -+ + -Dox. (1 µµµµg/ml) Cont./Chang Bcl-xL/Chang Cont./Chang Bcl-xL/Chang
A
B
Fig. 7. Effect of WT ERK2 overexpression in control cells or DN ERK2 overexpression in Bcl-xL-overexpressing cells on doxorubicin-induced release of mitochondrial cytochrome c. (A) Doxorubicin-induced release of mitochondrial cytochrome c is affected by intracellular ERK2 activity. Chang cells were transfected with the plasmid encoding WT ERK2 or pcDNA3 (0.1 µg). Bcl-xL/Chang cells were transfected with the DN ERK2 or pcDNA3 (0.3 µg). These transfected cells were further treated with or without 1 µg/ml doxorubicin for 24 h. Cells were fixed and immunoctochemistry was performed to detect cytochrome c and nuclei using anti-cytochrome c antibody and PI. Representative confocal images are shown (magnification 1200X). (B) Quantitative analysis of the cells whose mitochondrial cytochrome
c ωασ ρελεασεδ ιν Χηανγ χελλσ τρανσφεχτεδ ωιτη ∆Ν ΕΡΚ2 ορ Βχλ−ξΛ/Χηα
6. Doxorubicin-induced ERK2 activation is specifically mediated by Bcl-xL
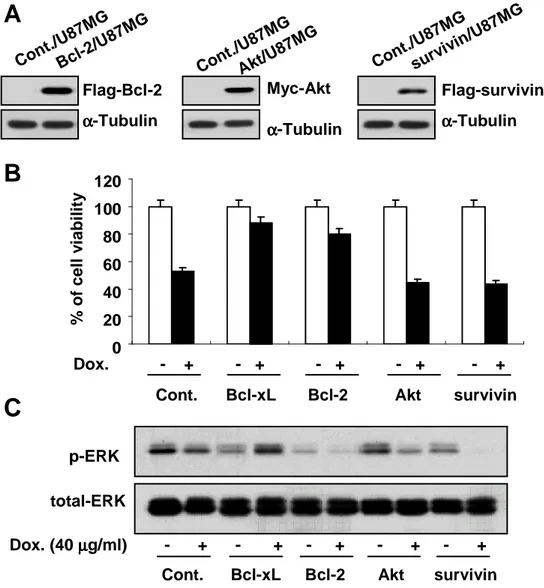
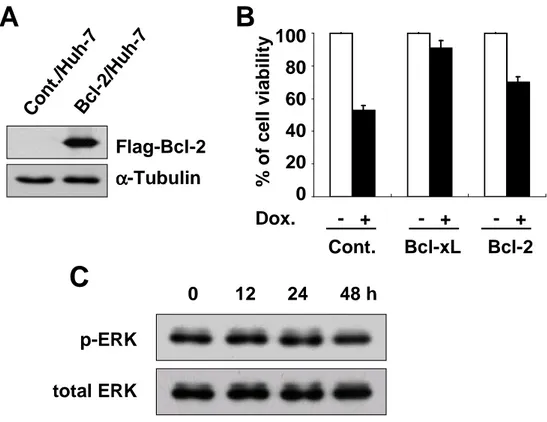
Next, we investigated whether other anti-apoptotic proteins also could block doxorubicin-induced apoptosis via ERK2 activation. We first examined the effect of the overexpression of Bcl-2, Akt or survivin on doxorubicin-induced apoptosis. Bcl-2 overexpression demonstrated significantly inhibited doxorubicin-induced cell death, although its anti-apoptotic effect was lesser than that of Bcl-xL (Fig. 8A and 8B). However, overexpression of Akt or survivin was unable to inhibit doxorubicin-induced cell death. We further examined whether Bcl-2 overexpression could elevate ERK2 activity in response to doxorubicin. While ERK2 was activated in Bcl-xL-overexpressing cells treated doxorubicin, it was not activated in Bcl-2-overexpressing cells (Fig. 8C). ERK2 was activated neither in Akt–Bcl-2-overexpressing cells nor in survivin-overexpressing cells. We further tested the effect of Bcl-2 overexpression on doxorubicin-induced apoptosis and ERK2 activity in Huh-7 cells. Bcl-2 overexpression in Huh-7 cells also demonstrated an anti-apoptotic effect on doxorubicin-induced apoptosis at lesser extent than Bcl-xL overexpression (Fig. 9A and 9B). Also in Huh-7 cell lines overexpressing Bcl-2, ERK2 was not activated following doxorubicin treatment (Fig. 9C). Taken together, doxorubicin-induced ERK2 activation is mediated by Bcl-xL, possibly contributing to strong anti-apoptotic function of Bcl-xL.
A
Flag-Bcl-2 α αα α-Tubulin Cont. /U87 MG Bcl-2 /U87 MG Cont. /U87 MG Akt/U 87MG Cont. /U87 MG survi vin/U 87MG Myc-Akt α αα α-Tubulin Flag-survivin α αα α-Tubulin 120 100 80 60 40 20 0 % o f c e ll v ia b il it y Dox. - + - + - + - + - + Cont. Bcl-xL Bcl-2 Akt survivinB
p-ERK total-ERK
Dox. (40 µµµµg/ml) - + - + - + - + - +
C
Cont. Bcl-xL Bcl-2 Akt survivin
Figure 8. Bcl-2 overexpression in U87MG cells blocks doxorubicin-induced apoptosis without accompanying activation of ERK2. (A) Stable U87MG cell lines overexpresing Flag-tagged Bcl-2 or survivin or Myc-tagged Akt were established and their expression was determined by Western blotting using anti-Flag or anti-Myc antibodies. (B) U87MG cell lines overexpressing Bcl-xL, Bcl-2, Akt, or survivin were treated with 40 µg/ml doxorubicin for 24 h and cellular viability was assessed using Live/Dead kit. (C) U87MG cell lines overexpressing Bcl-xL, Bcl-2, Akt, or survivin were treated with 40 µg/ml doxorubicin for 24 h or not treated. Protein levels of active ERK and total ERK were determined by Western blotting using phospho-sepcific ERK antibody and anti-ERK antibody.
Flag-Bcl-2 α αα α-Tubulin % o f c e ll v ia b il it y 100 80 60 40 20 0 Cont. Bcl-2 Dox. - + - + - + p-ERK total ERK Co nt. /Hu h-7 Bcl -2/H uh -7
A
0 12 24 48 h Bcl-xLB
C
Figure 9. ERK2 is not activated in Bcl-2-overexpressing Huh-7 cells treated with doxorubicin. (A) Expression of Bcl-2 in the stably transfected Huh-7 cells with Flag-tagged Bcl-2. Western blotting of Bcl-2 was performed in control Huh-7 cells and 2/Huh-7 cells using anti-Flag antibody. (B) Control Huh-7 cells and Bcl-2/Huh-7 cells were treated with 10 µg/ml doxorubicin for 24 h and cellular viability was measured using calcein-AM and EthD-1. (C) Bcl-2/Huh-7 cells were treated with 10 µg/ml doxorubicin for the indicated time periods and cell extracts were prepared for Western blotting to detect active ERK and total ERK protein levels.
B. Bcl-xL blocks apoptosis but not mitotic cell death induced by
doxorubicin
1. Bcl-xL overexpression blocks doxorubicin-induced apoptosis in Huh-7
hepatoma cells
While apoptosis was induced in Huh-7 cells treated with 10 µg/ml of doxorubicin, doxorubicin-induced apoptosis was blocked in Bcl-xL-overexpressing cells as described in part A (Fig 2).
2. Bcl-xL delays LD doxorubicin-induced mitotic cell death
Bcl-xL is well-known to be protective against apoptosis induced by various stimuli (Kirkin V et al., 2004). But, it has not been clarified whether Bcl-xL affects senescence or mitotic cell death induced by anticancer drugs of low doses. Our previous study demonstrated that LD doxorubicin induces mitotic cell death in human hepatoma cells. Therefore, we investigated whether Bcl-xL overexpression affects LD doxorubicin-induced mitotic cell death. First, we examined whether LD doxorubicin-induced changes in cellular morphologies are modulated by Bcl-xL overexpression. Huh-7 cells or Bcl-xL-overexpressing Huh-7 cells were treated with 50 ng/ml doxorubicin for the indicated time points and cells were stained with DAPI and Texas-Red-conjugated phalloidin to detect the nuclei and actin expression (Fig. 10). Cells were enlarged and multinucleation was significantly induced after treatment with LD doxorubicin in both Huh-7 and Bcl-xL-overexpressing cells.
Since SLP was observed in Huh-7 cells during the progression of LD doxorubicin-induced mitotic cell death, we investigated whether Bcl-xL overexpression could inhibit SLP. Huh-7 or Bcl-xL overexpressing Huh-7 cells were treated with 50 ng/ml doxorubicin for the indicated time periods and assay for SA-β-galactosidase activity, a biomarker for cellular senescence, were performed (Fig. 11). SA-β-gal activities in both cell lines were gradually increased following LD doxorubicin treatment in incubation time-dependent manner, although the increase in SA-β-gal activity was slightly delayed in Bcl-xL-overexpressing cells. These results suggest that Bcl-xL overexpression does not block the induction of SLP, a preceding event of LD doxorubicin-induced mitotic cell death. Next, we investigated whether Bcl-xL overexpression could inhibit LD doxorubicin-induced mitotic cell death. Cellular viability was slowly decreased in Huh-7 cells treated with 50 ng/ml doxorubicin, resulting in 50% at 9 days (Fig. 12). In Bcl-xL-overexpressing cells treated with 50 ng/ml doxorubicin, the viability was also decreased in time-dependent manner, although reduction of cellular viability in these cells was slightly delayed. Therefore, Bcl-xL overexpression cannot block LD doxorubicin-induced mitotic cell death.
3. Bcl-xL does not inhibit LD doxorubicin-induced aberrant cell cycle
progression and downregulation of cell cycle regulators.
Next, we examined the cell cycle progression of Huh-7 or Bcl-xL-overexpressing Huh-7 cells treated with LD doxorubicin, monitoring the changes in DNA contents by FACS analysis (Fig. 13A). In Huh-7 cells treated with LD doxorubicin, the G0/G1
cell populations were decreased at 1 day and then cells were transiently arrested at G2/M phase at 3 day. From this time, hyperploid cells (>4n DNA cells) appeared and gradually increased thereafter. While cellular viability was decreased by 50% at 9 days of doxorubicin treatment, the percentage of sub-G1 (apoptotic) population was not significantly increased, suggesting that the major mode of cell death by LD doxorubicin was not apoptosis. This aberrant cell cycle progression was also observed in Bcl-xL overexpressing cells treated with LD doxorubicin. We further examined the protein levels of several regulators of mitosis, Cdc2, Cdc25c, Cyclin A, Cyclin B, MAD2 and CENP-A in Huh-7 or Bcl-xL overexpressing Huh-7 cells treated with LD doxorubicin (Fig. 13B). All tested cell cycle regulators were downregulated in both cell lines treated with LD doxorubicin in time-dependent manner, suggesting that Bcl-xL overexpression does not inhibit LD doxorubicin-induced aberrant cell cycle progression.
4. Missegregation of chromosomes and loss in the integrity of nuclear
membranes are observed in both Huh-7 and Bcl-xL overexpressing Huh-7 cells
treated with LD doxorubicin
Next, we examined the changes of nuclear morphologies and microtubule structure during LD doxorubicin-induced mitotic cell death by immunocytochemistry using DAPI and anti-α-tubulin antibody (Fig. 14). In a few untreated Huh-7 or Bcl-xL-overexpressing Huh-7 cells, possibly cells undergoing mitosis, bipolar spindle structure and equally segregated chromosomes into opposite poles were observed.
However, cells with tri- or quadric-polar spindle structures were frequently observed both in Huh-7 and Bcl-xL-overexpressing cells after treatment with LD doxorubicin. These aberrant spindle structures may contribute to abnormal mitosis via missegregation of chromosomes and subsequent formation of anueploid cells. Next, we analyzed the changes in the integrity of nuclear membrane by LD doxorubicin treatment. Huh-7 or Bcl-xL overexpressing Huh-7 cells were treated LD doxorubicin for the indicated time periods were stained with the specific antibody for Lamin B, a component of nuclear envelope (Fig.15A). Untreated Huh-7 or Bcl-xL-overexpressing Huh-7 cells had intact nuclear membrane structure. However, disrupted nuclear membrane boundaries were frequently observed both types of cells treated with LD doxorubicin. Lamin B protein levels were also downregulated in both cells (Fig. 15B). These results suggest that Bcl-xL overexpression does not block aberrant formation of spindle structures and loss of nuclear membrane integrity induced by LD doxorubicin.
Control
3 days
6 days
9 days
Nuclei
Actin
Nuclei
Actin
Cont./
Huh-7
Bcl-xL/
Huh-7
Fig. 10. Bcl-xL did not block LD doxorubicin-induced multinucleation. Huh-7 and Bcl-xL-overexpressing Huh-7 cells were treated with LD doxorubicin for the indicated time periods. Immunocytochemistry using DAPI and Texas-Red-conjugated phalloidin was performed to detect the changes in the nuclei and actin structure. Cells were visualized by confocal laser microscopy (magnification 1200X).
100
80
60
40
20
0
0 1 2 3 4 5 6 9 12 days
%
o
f
S
A
-
ββββ
-g
a
l
–
(+
)
c
e
ll
s
Cont./Huh-7
Bcl-xL/Huh-7
Cont./Huh-7
Bcl-xL/Huh-7
A
B
Control
3 days
6 days
Cont./
Huh-7
Bcl-xL/
Huh-7
Fig. 11. Bcl-xL delays LD doxorubicin-induce senescence. (A) Expression of SA-β-galactosidase. Huh-7 and Bcl-xL overexpressing Huh-7 cells were treated with 50 ng/ml of doxorubicin for the indicated time periods and then SA-β-gal assay was performed. Representative pictures are shown. (B) Quantitation of SA-β-gal-positive cells.
A
- Dox
+ Dox
Cont./
Huh-7
Bcl-xL/
Huh-7
100
80
60
40
20
0
%
o
f
c
e
ll
v
ia
b
il
it
y
0 2 4 6 9 12 15 days
B
Cont./Huh-7
Bcl-xL/Huh-7
Cont./Huh-7
Bcl-xL/Huh-7
Fig. 12. Bcl-xL delays but does not block LD doxorubicin-induced mitotic cell death in Huh-7 cells. (A) Bcl-xL did not block LD doxorubicin-induced enlargement of cells. Huh-7 cells or Bcl-xL-overexpressing Huh-7 cells were treated with 50 ng/ml of doxorubicin for 5 days. Representative pictures taken under phase contrast microscope are shown. (B) Bcl-xL overexpression slightly delayed but did not block doxorubicin-induced mitotic cell death. Cells were treated with 50 ng/ml of doxorubicin for the indicated time periods and cellular viability was measured using calcein-AM and EhtD-1.
Cdc2 Cdc25c Cyclin A Cyclin B MAD2 CENP-A
A
Cont./ Huh-7 Bcl-xL/ Huh-7Control 3 days 6 days 9 days
B
Cont./Huh-7 Bcl-xL/Huh-7
Fig. 13. Bcl-xL does not inhibit aberrant cell cycle progression and downregulation of cell cycle regulators. (A) Huh-7 and Bcl-xL-overexpressing Huh-7 cells were treated with LD doxorubicin for the indicated time periods and the profile of cell cycle progression was analyzed by FACS. (B) Cells were treated with LD doxorubicin for the indicated time periods and Western blotting of the cell cycle regulators was performed.
3 days
Control
αααα
-tubulin
Nuclei
Cont./
Huh-7
Nuclei
Bcl-xL/
Huh-7
αααα
-tubulin
Fig. 14. Missegregation of chromosomes is observed not only in Huh-7 cells but also in Bcl-xL-overexpressing Huh-7 cells treated with LD doxorubicin. Huh-7 and Blc-xL-overexpressing Huh-7 cells were treated with or without LD doxorubicin and cells were stained with anti-α-tubulin antibody and DAPI. Cells were visualized in confocal laser microscopy (magnification 1200X).
Lamin B
Nuclei
Control 3 days 6 days 9 days
Cont./
Huh-7
Lamin B
Nuclei
Bcl-xL/
Huh-7
A
Cont./
Huh-7
Bcl-xL/
Huh-7
0 1 2 3 6 9 12 15 days
B
Fig. 15. Loss in the integrity of nuclear membrane is observed in both Huh-7 and Bcl-xL-overexpressing Huh-7 cells treated with LD doxorubicin. (A) Huh-7 and Bcl-xL overexpressing Huh-7 cells were treated with LD doxorubicin for the indicated time periods and stained with anti-Lamin B antibody and DAPI. Cells were visualized in confocal laser microscopy (magnification 1200X). Arrow shows the loss nuclear membrane integrity. (B) Western blotting of Lamin B in control cells and Bcl-xL-overexpressing Huh-7 cells treated with LD doxorubicin.
5. Bcl-xL overexpression does not inhibit loss of mitochondrial function and
release of inner-mitochondrial proteins into cytosol in LD doxorubicin-induced
mitotic cell death.
Bcl-xL functions to prevent the loss of outer membrane integrity by preventing both membrane hyperpolarization and mitochondrial swelling in response to various apoptotic stimuli (Kirkin V et al., 2004). However, it has not been clarified whether these changes of mitochondria observed during apoptosis are also occurred during mitotic cell death. To investigate whether mitochondrial function is lost during LD doxorubicin-induced mitotic cell death, we first examined the changes of mitochondrial membrane potential (MMP) of 7 or Bcl-xL-overexpressing Huh-7 cells using Rhodamine 123 and JC-1, mitochondrial specific dyes (Fig. 16). While cells with intact MMP are usually stained with Rhoeamine 123 strongly, cells whose MMP is lost are stained with it weakly. At 2 day of LD doxorubicin treatment, MMP was transiently increased in Huh-7 cells but significantly decreased thereafter. Bcl-xL-overexpressing cells had a slightly stronger staining pattern of Rhaodamine 123. Transient increase in Rhodamine 123 staining was slightly delayed but its subsequent reduction was not affected in Bcl-xL-overexpressing cells treated with LD doxorubicin. Similar results were obtained using JC-1 day. JC-1 dye emits green signal upon reduction in MMP (see the Materials and Methods). Percentages of the cells with green fluorescence were increased in time-dependent manner in Huh-7 or Bcl-xL-overexpressing Huh-7 cells. Next, we measured intracellular ATP levels. In both Huh-7 and Bcl-xL-overexpressing Huh-7 cells treated with LD doxorubicin,
ATP levels were significantly dropped (Fig. 17). When cellular MMP is lost, inner-mitochondrial proteins are released into cytosol. Therefore, we tested the release of inner-mitochodnril proteins, cytochrome c and Smac, by immunocytochemistry. Puntated appearance of cytochrome c or Smac represents its mitochondrial localization and diffuse form represents release into cytosol. Cytochrome c was localized in mitochondria in Huh-7 and Bcl-xL-overexpressing Huh-7 cells untreated with doxorubicin. However, in both types of LD doxorubicin-treated cells, mitochondrial cytochrome c was released into cytosol (Fig. 18A). Subcellular fractionation and the subsequent Western blotting analysis also demonstrated the increase in the amount of cytosolic cytochrome c (Fig. 18B). Similar results were observed with Smac (Fig. 20). We compared the mitochondrial changes during LD doxorubicin-induced mitotic cell death with those during HD doxorubicin-induced apoptosis (Fig. 20 and 21). Bcl-xL overexpression effectively blocked the loss of MMP and ATP depletion by HD doxorubicin. HD doxorubicin-induced release of mitochondrial cytochrome c and Smac was also inhibited by Bcl-xL overexpression. These results suggest that Bcl-xL overexpression does not inhibit LD doxorubicin-induced loss of mitochondrial function, whereas it significantly blocks HD doxorubicin-induced loss of mitochondrial function.
B
Con 3 days 6 days 9 days Con 2 days 4 days 6 daysA
Cont./ Huh-7 Bcl-xL/ Huh-7 Cont./ Huh-7 Bcl-xL/ Huh-7Fig. 16. Bcl-xL does not block LD doxorubicin-induced loss of mitochondrial membrane potential. (A) Huh-7 cells and Bcl-xL overexpressing Huh-7 cells were treated with LD doxorubicin for the indicated time periods and then incubated with Rhodamine 123. Cells were visualized in fluorescent microscopy. (B) Cells were treated with LD doxorubicin and then incubated with JC-1 and analyzed by FACS.
100
80
60
40
20
0
A
T
P
c
o
n
c
e
n
tr
a
ti
o
n
(
%
)
0 3 6 9 12 days
Cont./Huh-7
Bcl-xL/Huh-7
Cont./Huh-7
Bcl-xL/Huh-7
Fig. 17. Bcl-xL slightly delays doxorubicin-induced loss of intracellular ATP levels. ATP levels were measured in Huh-7 or Bcl-xL-overexpressing Huh-7 cells treated with LD doxorubicin for the indicated time periods.
0 3 6 9 12 days
Cyt. c
Nuclei
Control 3 days 6 days 9 days
Cont./
Huh-7
Nuclei
Bcl-xL/
Huh-7
A
B
Cyt. c
Cont./
Huh-7
Bcl-xL/
Huh-7
Fig. 18. Release of cytochrome c is observed not only in Huh-7 cells but also in Bcl-xL overexpressing cells treated with LD doxorubicin. (A) Huh-7 and Bcl-xL overexpressing Huh-7 cells were treated with LD doxorubicin for the indicated time periods and stained with anti-cytochrome c antibody and DAPI. Cells were visualized in confocal laser microscopy (magnification 1200X). (B) Subcellular fractionation and Western blotting of cytochrome c. After subcellular fractionation, equal amount of cytosolic fraction (10 µg) was loaded on SDS-polyacrylamide gel for Western blotting.
Smac
Nuclei
Cont./
Huh-7
Nuclei
Bcl-xL/
Huh-7
Smac
Control 3 days 6 days 9 days
0 3 6 9 12 days
Cont./
Huh-7
Bcl-xL/
Huh-7
B
A
Fig. 19. Release of Smac is observed not only in Huh-7 cells but also in Bcl-xL overexpressing cells treated with LD doxorubicin. (A) Huh-7 and Bcl-xL overexpressing Huh-7 cells were treated with LD doxorubicin for the indicated time periods and stained with anti-Smac antibody and DAPI. Cells were visualized in confocal laser microscopy (magnification 1200X). (B) Subcellular fractionation and Western blotting of Smac. After subcellular fractionation, equal amount of cytosolic fraction (10 µg) was loaded on SDS-polyacrylamide gel for Western blotting.
120
100
80
60
40
20
0
A
T
P
c
o
n
c
e
n
tr
a
ti
o
n
(
%
)
60
50
40
30
20
10
0
%
o
f
g
re
e
n
c
e
ll
s
0 48 72 h
0 12 24 48 h
A
B
Cont./Huh-7
Bcl-xL/Huh-7
Cont./Huh-7
Bcl-xL/Huh-7
Cont./Huh-7
Bcl-xL/Huh-7
Cont./Huh-7
Bcl-xL/Huh-7
Fig. 20. Bcl-xL blocks HD doxorubicin-induced loss of mitochondrial membrane potential and ATP levels. (A) Huh-7 and Bcl-xL overexpressing Huh-7 cells treated with HD doxorubicin and then incubated with JC-1 and analyzed by FACS. (B) Cellular energy status of Huh-7 and Bcl-xL overexpressing Huh-7 cells treated with HD doxorubicin.
A
Smac Nuclei Cyt. c Nuclei 0 4 8 12 24 h Cont./ Huh-7 Bcl-xL/ Huh-7 Cont./ Huh-7 Bcl-xL/ Huh-7B
C
D
Cont./ Huh-7 Bcl-xL/ Huh-7 0 4 8 12 24 h Cont./ Huh-7 Bcl-xL/ Huh-7Fig. 21. Bcl-xL blocks HD doxorubicin-induced release of mitochondrial cytochrome c and Smac into cytosol. (A) Cells were treated with doxorubicin for 24 h and stained with anti-cytochrome c antibody and PI. Cells were visualized under confocal microscopy (magnification 1200X). (B) Subcellular fraction and Western blotting by cytochrome c. After subcellular fractionation, equal amount of cytosolic fraction (10 µg) wasloaded on SDS-polyacrylamide gel for Western blotting. (C) Cells were treated with or without doxorubicin for 24 h and stained with anti-Smac antibody and PI. Cells were visualized under confocal microscopy (magnification 1200X). (D) Subcellular fractionation and Western blotting of cytosolic Smac.
6. Not only Bcl-xL but also Bcl-2 does not block mitotic cell death
Next, we compared the effect of Bcl-xL overexpression on HD doxorubicin-induced apoptosis and LD doxorubicin-induced mitotic cell death using Chang cells. Bcl-xL overexpression in Chang cells significantly inhibited HD (1 µg/ml) doxorubicin-induced apoptosis (Fig 22A), as expected. However, it did not inhibit LD (25 ng/ml) doxorubicin-induced multinucleation and subsequent mitotic cell death (Fig. 22B and 22C). Next, we investigated whether Bcl-2, an anti-apoptotic protein with similar function of Bcl-xL (Kirkin V et al., 2004), affects LD doxorubicin-induced mitotic cell death. We established the stable Huh-7 cell lines overexpressing Bcl-2 and cellular viability was measured after treatment with 50 ng/ml of doxorubicin (Fig. 23). Cellular viability was decreased gradually by 50% at 9day of LD doxorubicin treatment both in Huh-7 and Bcl-2 overexpressing Huh-7 cells. Therefore, Bcl-xL overexpression as well as Bcl-2 overexpression does not inhibit LD doxorubicin-induced mitotic cell death.
7. Bcl-xL does not enhance long-term survival of the cells treated with LD
doxorubicin
We next analyzed the effect of LD doxorubicin on long-term survival by clonogenic assay. After treatment with LD doxorubicin for the indicated time periods, colonies were formed neither in Huh-7 nor in Bcl-xL overexpressing Huh-7 cells, compared with the untreated control group (Fig. 24). In contrast, colony formation in Bcl-xL overexpressing Huh-7 cells treated with HD doxorubicin for short time was more
increased than in control Huh-7 cells treated with HD doxorubicin for the same period (data not shown). These results demonstrate that Bcl-xL overexpression does not enhance long-term survival following treatment with LD doxorubicin.
100 80 60 40 20 0 % o f c e ll v ia b il it y 100 80 60 40 20 0 % o f c e ll v ia b il ity Cont./Chang Bcl-xL/Chang Cont./Chang Bcl-xL/Chang - Dox + Dox 0 3 6 9 12 days
A
C
B
control 3 days control 3 days
Cont./Chang Bcl-xL/Chang Nuclei Actin Cont./Chang Bcl-xL/Chang Cont./Chang Bcl-xL/Chang
Fig. 22. Bcl-xL overexpression in Chang cells blocks HD doxorubicin-induced apoptosis but not LD doxorubicin-induced mitotic cell death. (A) Bcl-xL overexpression in Chang cells inhibits HD (1 µg/ml) doxorubicin-induced apoptosis. (B) Bcl-xL overexpression in Chang cells did not block LD doxorubicin-induced multinucleation. (C) Bcl-xL overexpression did not block LD doxorubicin-induced mitotic cell death.
Cont./Huh-7
Bcl-2/Huh-7
Cont./Huh-7
Bcl-2/Huh-7
0 2 4 6 9 12 15 days
100
80
60
40
20
0
- Dox
+ Dox
Cont./
Huh-7
Bcl-2/
Huh-7
%
o
f
c
e
ll
v
ia
b
il
it
y
A
B
Fig. 23. 2 does not block LD doxorubicin-induced mitotic cell death. (A) Bcl-2 overexpression in Huh-7 cells did not block LD doxorubicin-induced enlargement of cells. Huh-7 cells and Bcl-2-overexpressing Huh-7 cells were treated with 50 ng/ml of doxorubicin for 5 days. Representative pictures taken under phase contrast microscope are shown. (B) Cells were treated with 50 ng/ml of doxorubicin for the indicated time periods and cellular viability was measured using calcein-AM and EthD-1.
0 h 12 h 24 h 48 h
Cont./
Huh-7
Bcl-xL/
Huh-7
Fig. 24. Bcl-xL does not enhance long-term survival of the cells treated with LD doxorubicin. Huh-7 or Bcl-xL overexpressing Huh-7 cells were treated with LD doxorubicin for the indicated time periods and further incubated in fresh medium without doxorubicin for 12 days. Colony forming assay using crystal violet was performed.
IV. DISCUSSION
A. Bcl-xL-mediated ERK2 activation plays a critical role in the inhibition of
doxorubicin-induced apoptosis
Bcl-xL is one of the representative anti-apoptotic molecules against various cytotoxic stimuli and it is widely overexpressed in various tumor cells (Kirkin V et
al., 2004). Bcl-xL has been demonstrated to exert its anti-apoptotic function through
the prevention of mitochondrial changes such as mitochondrial membrane potential, cytochrome c release, and the production of reactive oxygen species (Kirkin V et al., 2004). However, it is not well characterized whether the anti-apoptotic function of Bcl-xL in response to cytotoxic stimuli is associated with modulation of the activities of signaling molecules. We demonstrate here for the first time that ERK2 activated in Bcl-xL-overexpressing cells treated with doxorubicin significantly contribute to the death blocking effect of Bcl-xL. ERK2 activation in response to doxorubicin was observed not only in overexpressing Huh-7 cells (Fig. 3) but also in Bcl-xL-overexpressing Chang or U87MG cells (Fig. 4), suggesting that this response is generally induced irrespective of cell types and ERK2 activated NF-κB. We tested whether overexpression of other anti-apoptotic molecules also can inhibit doxorubicin-induced apoptosis through ERK2 activation (Fig. 8). Although overexpression of Bcl-2 but not Akt or survivin, inhibited doxorubicin-induced
apoptosis at lesser extent than Bcl-xL overexpression, ERK2 activation was not observed in Bcl-2 overexpressing cells treated with doxorubicin. These results suggest that ERK2 activation in response to doxorubicin is specifically mediated by Bcl-xL contributing to strong anti-apoptotic activity of Bcl-xL.
Activation of the ERK cascade has been generally known to lead cell proliferation or survival (Chang F et al., 2004), although pro-apoptotic role of ERK has been reported in cisplatin–induced apoptosis in human cervical carcinoma HeLa cells (Wang X et al., 2000) or radiation–induced cell death in HSP25-overexpressed L929 cells (Cho HN et al., 2002). Therefore, ERK may play an anti-apoptotic or pro-apoptotic role depending on the cytotoxic stimuli or cellular contexts. ERK2 has been generally postulated to have proproliferative effects, while ERK1 has antiproliferative effects (Chang F et al., 2004). However, little is known how different in vivo targets of ERK1 and ERK2 are. In our study, mainly ERK2 was activated in Bcl-xL-overexpressing treated with doxorubicin (Fig. 3 and 4). Functional significance of ERK2 activation in Bcl-xL-mediated blocking of doxorubicin-induced apoptosis was confirmed by the following lines of evidences. First, overexpression of DN ERK2 sensitized doxorubicin-induced apoptosis in Bcl-xL overexpressing Chang cells (Fig. 5). Second, overexpression of ERK2 in Chang cells alleviated doxorubicin-induced apoptosis (Fig. 6).
MEK is known to be a representative upstream kinase of ERK (Chang F et al., 2004). However, in our study the activity of MEK, a well-known upstream activator of ERK, was not increased in Bcl-xL-overexpressing Huh-7 cells treated with