이학 석사학위 논문
CDDO-ME inducesapoptosis
in breast cancer cells
via Ca
2+
influx-mediated dilation of
endoplasmic reticulum and c-FLIP
downregulation
아 주 대 학 교 대 학 원
의생명과학과/종양생물학전공
CDDO-ME induces apoptosis
in breast cancer cells via Ca
2+influx-mediated
dilation of endoplasmic reticulum
and c-FLIP downregulation
by
SooAh Jeong
A Dissertation Submitted to The Graduate School of Ajou University in Partial Fulfillment of the Requirements
for the Degree of
Master of Biomedical Sciences
Supervised by
Kyeong Sook Choi, Ph.D.
Major in Cancer Biology
Department of Biomedical Sciences
February, 2014
This certifies that the dissertation
of SooAh Jeong is approved.
SUPERVISORY COMMITTEE
심사위원장조혜성인
심사위원최경숙인
심사위원김유선인
The Graduate School, Ajou University
December, 19th, 2014
i
-Abstract -
CDDO-ME induces apoptosis in breast cancer cells
via Ca
2+influx-mediated dilation of endoplasmic reticulum
and c-FLIP downregulation
Oleanolic acid-derived synthetic triterpenoids are a promising new class of compounds with antitumorigenic activity against various types of cancer cells. In this study, we show that methyl-2-cyano-3, 12-dioxooleana-1, 9(11)-dien-28-oate (CDDO-ME)triggered paraptosis-like cellular vacuolation, which was mainly derived from the dilation of the endoplasmic reticulum, but finally killed breast cancer cells via apoptosis.We found that the increase in intracellular Ca2+ levelspreceded CDDO-ME-induced vacuolation and cell death. In addition, this increase was accompanied by the increase in reactive oxygen species (ROS) levels. Pretreatment with Ca2+ chelator, BAPTA, BAPTA-AM or antioxidant, N-acetylcysteine effectively blocked CDDO-ME-induced vacuolation and cell death, suggesting that Ca2+ influx and ROS generation are initial critical signals for CDDO-ME-induced anti-cancer effect. Moreover, CDDO-ME markedly reduced the protein levels of c-FLIPL. Overexpression of c-FLIPL did not affect CDDO-ME-induced vacuolation, but significantly attenuated CDDO-ME-induced
ii
cell death. Interestingly,CDDO-ME increased intracellular Ca2+ and ROS levels at much lower doses, compared to CDDO and CDDO rather increased the protein levels of c-FLIPL via its proteasome inhibitory activity. Taken together, our results clearly show that both intracellular Ca2+ influx, ROS generation and c-FLIP downregulation critically contribute to the potent anti-cancer effect of CDDO-ME in breast cancer cells.
iii
TABLE OF CONTENS
- Abstract - ... i TABLE OF CONTENS ...iii LIST OF FIGURES ... v I. INTRODUCTION ... 1 II. MATERIALS AND METHODS... 6 A. Chemicals and antibodies ... 6 B. Cell culture of various cancer cell lines ... 7 C. Measurement of cell viability ... 7 D. Western blotting ... 7 E. Immunocytochemistry ... 8 F. Establishment of the stable cell lines in the fluorescence specifically mitochondria or endoplasmic reticulum ... 9 G. Measurement of ROS and mitochondrial superoxide anion ... 9 H. Measurement of cytosolic and mitochondrial Ca²⁺ levels ... 9 I. shRNA-mediated knockdown of proteins ... 10 J. Transmission electron microscopy ... 10 K. Reverse transcription–PCR analysis ... 11 L. Statistical analysis ... 12 III. RESULTS ... 13 1. CDDO-ME demonstrates a potent anti-cancer effect on breast cancer cells. ... 13 2. CDDO-ME induces paraptosis-like cellular vacuolation prior to morphologies features of apoptosis in breast cancer cells ... 17
iv
3. Ca2+ influx is crucial for CDDO-ME-induced vacuolation and subsequent apoptotic cell
death ... 40 4. Cross-modulation between Ca2+ influx and ROS generation critically contributes to
CDDO-ME-induced vacuolation and subsequent apoptosis ... 51 5. c-FLIPL downregulation plays a critical role in CDDO-ME-induced apoptotic cell death,
but not in vacuolation... 58 6. Higher increase in intracellular Ca2+ and ROS levels as well as c-FLIP downregulation
may contribute to a more potent anti-cancer effect of CDDO-ME, compared to CDDO 69 IV. DISCUSSION ... 73 V. REFERENCES ... 79 - 국문요약 - ... 90
v
LIST OF FIGURES
Figure 1. Structures of CDDO and CDDO-ME ... 14 Figure 2. Effects of CDDO and CDDO-ME on the viability of various breast cancer cells. .. 15 Figure 3. Dose-dependent effects of CDDO-ME and CDDO on the long-term survival of MDA-MB 435S cells. ... 16 Figure 4. CDDO-ME induces extensive dilation prior to apoptotic cell death in MDA-MB 435S cells. ... 23 Figure 5. Knockdown of ATG5, beclin-1, LAMP2 does not block cell death and vacuolation induced by CDDO-ME. ... 24 Figure 6. Effects of autophagy inhibitors on CDDO-ME-induced vacuolation and cell death…. ... 25 Figure 7. Autophagy-related proteins, LC3B and p62, were accumulated in MDA-MB 435S cells treated with CDDO-ME. ... 26 Figure 8. CDDO-ME induces dilation of the ER and mitochondrial fragmentation.. ... 27 Figure 9. Morphological changes of the ER and mitochondria in CDDO-ME-treated breast cancer cells. ... 28 Figure 10. CDDO-ME increases the protein levels of ER stress-associated proteins in MDA-MB 435S cells. ... 29 Figure 11. CDDO-ME induces ER stress in MDA-MB 435S cells. ... 30 Figure 12. Electron microscopic observation of MDA-MB 435S cells treated with CDDO-ME. ... 31
vi
Figure 13. Electron microscopic images of MDA-MB 435S cells treated with curcumin or celastrol. ... 32 Figure 14. Cycloheximide, a translation inhibitor, does not block CDDO-ME-induced cell death and vacuolation. ... 33 Figure 15. CDDO-ME-induced ER dilation is not blocked by cycloheximide. ... 34 Figure 16. Change in protein expression of Alix. ... 35 Figure 17. Activation of caspases in MDA-MB 435S cells treated with CDDO-ME... 36 Figure 18. Inhibition of caspase activation is critically involved in CDDO-ME-induced cell death.. ... 37 Figure 19. CDDO-ME induces chromatin condensation and release of cytochrome c... 38 Figure 20. Effect of CDDO-ME on MDA-MB 435S cell cycle progression.. ... 39 Figure 21. CDDO-ME increases intracellular Ca2+ levels.. ... 43
Figure 22. CDDO-ME does not affect mitochondrial Ca2+ levels. ... 44
Figure 23. Effect of Several Ca2+ inhibitors on MDA-MB 435S cells treated with CDDO-ME..
... 45 Figure 24. Dilation of the ER is blocked by BAPTA, BAPTA-AM and EGTA. ... 46 Figure 25. Mitochondrial fragmentation is not associated with Ca2+ influx. ... 47
Figure 26. PARP cleavage is blocked by BAPTA-AM plus BAPTA in CDDO-ME-treated cells. ... 48 Figure 27. CDDO-ME increases intracellular Ca2+ levels in other breast cancer cells. ... 49
Figure 28. BAPTA plus BAPTA-AM block CDDO-ME-induced cell death in other breast cancer cells. ... 50
vii
Figure 29. CDDO-ME induces ROS generation in breast cancer cells... 53 Figure 30. Antioxidants block CDDO-ME-induced cell death. ... 54 Figure 31. CDDO-ME treatment had no effect on mitochondrial ROS generation. ... 55 Figure 32. Antioxidants blocked dilation of the ER induced by CDDO-ME.. ... 56 Figure 33. Cross-modulation between Ca2+ and ROS generation... 57
Figure 34. Change in protein levels of caspase antagonists in MDA-MB 435S cells treated with CDDO-ME. ... 61 Figure 35. CDDO-ME downregulates c-FLIPL in breast cancer cells.. ... 62
Figure 36. CDDO-ME modulates c-FLIPL in both transcriptional and post-transcriptional level.
... 63 Figure 37. CDDO-ME downregulates c-FLIPL expression by proteasome system. ... 64
Figure 38.c-FLIP downregulation is critical for apoptotic cell death but not for vacuolation by CDDO-ME.. ... 66 Figure 39. Overexpression of c-FLIP has similar effects to z-VAD-fmk. ... 67 Figure 40.c-FLIP downregulation is not associated with increase of both intracellular Ca2+ and
ROS levels.. ... 68 Figure 41.A more potent anti-cancer effect of CDDO-ME than that of CDDO may be due to enhanced Ca2+ influx, ROS generation. ... 70
Figure 42. The protein levels of c-FLIPL were accumulated by CDDO. ... 71
1
I. INTRODUCTION
Breast cancer is the most common cancer in woman and one of the leading causes of cancer death for women.Despite improvement in treatment options, the women who died from breast cancer in the worldare more than 450,000 anually (Garcia Met al., 2007). The rate of breast cancer has increased rapidly and the mortality of breast cancer patients has remained stable (Smigal C et al., 2006). Drug resistance remains the major source of treatment failure in women with breast cancer.Among various cancer therapies, such aschemotherapy,lumpectomy,mastectomy and radiation therapy(Early Breast Cancer Trialists’ Collaborative Group et al., 2006; Clarke M et al., 2005),combined with surgery and chemotherapy, radiotherapy continues to be a powerful toolin the treatment of breast cancer (Chavez KJet al., 2010).
Many scientists are currently seeking new anticancer agents with better efficiency and fewer side effects. The novel synthetic triterpenoid meth
yl-2-cyano-3,12-dioxoolean-1,9-dien-28-oate (CDDO-ME) holds promise as a cancer therapeutic agent and is currently being tested in phase II clinical trials (Wang YY et al., 2014). CDDO-ME potently induces apoptosis of cancer cells and exhibits antitumor activity in diverse types of tumor cell lines, including osteosarcoma, leukemia, prostate, lung cancer cells (Ryu K et al., 2010; Samudio I et al., 2008; Liu Y et al., 2012; Zou W
et al., 2008).In vivocancer preventive effects of CDDO-ME have been established in
various mouse models of breast cancer (Tran K et al., 2012; Kim EH et al., 2012; Liby K et al., 2008), suggesting that CDDO-ME may have therapeutic potential in breast
2
cancer therapy. Several reports have shown that generation of reactive oxygen species (ROS) is critically involved in the anti-proliferative and apoptosis-inducing activity of CDDO-ME in various cancer cells (Gao X et al., 2011; Deeb D et al., 2012). However, the death-inducing effect of CDDO-ME and its underlying mechanism has not been clarified yet. In this study, we show for the first time that breast cancer cells treated with CDDO-ME undergo extensive ER-derived vacuolation and finally culminate in apoptosis.
Cell death has two types: programmed cell death (PCD) and passive cell death. The PCD mediated by an intracellular program and has been classified morphologically into three main types: Apoptotic cell death (type 1 PCD), autophagic cell death (type 2 PCD), cytoplasmic cell death (type 3 PCD) (Sperandio S et al., 2004). Apoptosis may occur in multicellular organisms (Greenet al., 2011). Biochemical events lead to characteristic morphologyand death. Apoptotic morphology include chromosomal DNA fragmentation, chromatin condensation, nuclear fragmentation, cell shrinkage and blebbing. Also, cytochrome c is released from mitochondria and the sub-G1 portion increased. This cell death is blocked by pan-caspase inhibitor, z-VAD-fmk. Autophagy is characterized by the formation of large vacuoles that eat away organelles in a specific sequence prior to the destruction of the nucleus (Bursch W et al.,2000). During autophagy,bulk cytoplasmic contents, abnormal protein aggregates, and excess or damaged organelles were degraded by the autophagosomal-lysosomal fusion mechanism. Recently, called "non-apoptotic programmed cell-death (type 3)" can also function as the main type of PCD.Paraptosis(from para= next to or related to, and
3
apoptosis), a type of non-apoptotic cell death, has been reported by Sperandio S
(Sperandio Set al., 2000) and induced extensive cytoplasmic vacuolization, swelling of endoplasmic reticulum (ER) and/or mitochondria, without the characteristic apoptotic features of pyknosis, DNA fragmentation or caspase activation (Sperandio S et al., 2000; Wyllie AH et al., 2001).Paraptosis is also characterized by a requirement for new gene transcription and translation (Sperandio S et al., 2004). Various stimuli, such as curcumin and celastrol, were shown to induce paraptosis (Yoon MJ et al., 2010; Yoon MJ and Lee AR et al., 2014). Recent reports have shown that paraptosis can be inhibited by the expression of AIP-1/Alix (Sperandio S et al., 2004; Valamanesh F et al., 2007).
Ca2+ is essential for cell physiology. Movement of Ca2+ into and out of the cytoplasm functions as a key signal for many cellular processes, such as proliferation, differentiation and cell death including apoptosis.Ca2+ can act in signal transduction resulting from activation of ion channels, the plasma membrane calcium pump (PMCA), the endoplasmic reticulum Ca2+-ATPase (SERCA) and the mitochondrial Ca2+ transport (MCU) (Pimentel AA and Benaim Get al., 2012).The ER is well known to be a major reservoir of intracellular Ca2+ (Pivovarova NB et al., 2002). It serves as an intracellular Ca2+ storesthrough the release and/or uptake of Ca2+(Maggio Net al. 2014).ER Ca2+ is regulated by two major Ca2+ release receptors: (1) Ryanodine receptors (RyR), which amplify local cytosolic Ca2+-transients (Rose and Konnerthet al., 2001; Fill and Copelloet al., 2002, Meissner et al., 2002; Bouchard et al., 2003) (2) Inositol 1,4,5 triphosphate receptors (IP3R) to release Ca2+ from intracellular stores (Bezprozvanny and Ehrlichet al., 1995; Mikoshibaet al., 1997, Taylor and Laudeet al., 2002; Ribeiro et
4
al., 2010). Although many Ca2+ signaling pathway are well-known, the relationship between the dilation of the ER and Ca2+ flux is unclear.
Ectopic expression of c-FLIP variants decreased apoptosis caused by death ligands and anticancer agents (Kin Y et al., 2002, Safa AR et al., 2008), indicating that overexpression of these proteins may cause resistance to multiple anticancer drugs. c-FLIP is expressed as long (c-c-FLIPL), short (c-FLIPS), and c-FLIPR splice variants in human cells. c-FLIP binds to FADD and/or caspase-8 or -10 in a ligdependent and-independent fashion, which in turn prevents death-inducing signaling complex (DISC) formation and subsequent activation of the caspase cascade (Safa AR et al., 2012; Kataoka T. et al., 2003; Wajant H. et al., 2003; Irmler M. et al., 1997; Kim Y et al., 2002). Elevated c-FLIP levels are observed in various types of cancer cells and higher c-FLIP levels were shown to correlate with a more aggressive tumor (Poukkula M et
al.,2005). Downregulation of c-FLIP has been shown to restore apoptosis triggered by cytokines and various chemotherapeutic agents. Hence, c-FLIP is an important target for cancer therapy. In addition, the levels of c‑FLIP including both FLIPL, FLIPS and FLIPR are regulated by ubiquitin/proteasome‑mediated degradation (Kim Y et al., 2002; Chang L et al., 2006; Poukkula M et al., 2005). It has been reported that elevated c‑FLIP expression protects cells from death receptor‑mediated apoptosis in various cell types, whereas downregulation of c‑FLIP by chemicals or siRNA sensitizes cells to death receptor‑mediated apoptosis.
Here, we showed that CDDO-ME induced paraptosis-like vacuolation, which was mainly derived from the dilation of the endoplasmic reticulum, but finally killed breast
5
cancer cells via apoptosis. CDDO-ME increases not only Ca2+ levels but also total ROS levels as the critical signal.It effectively induces apoptotic cell death via ER dilation.In addition, CDDO-ME markedly reduced the protein levels of c-FLIPL,which is involved in cell death but not in vacuolation induced by CDDO-ME. Taken together, our results clearly show that both intracellular Ca2+ influx, ROS generation and c-FLIP downregulation critically contribute to the potent anti-cancer effect of CDDO-ME in breast cancer cells.
6
II. MATERIALS AND METHODS
A. Chemicals and antibodies
CDDO-ME and CDDO were obtained from Cayman Chemical(Ann arbor, MI), N-acetylcysteine (NAC), reduced glutathione (GSH), ethylene glycol tetraacetic acid (EGTA), bis(o-aminophenoxy)ethane-N,N,N’N’-tetraacetic acid (BAPTA), 1,2-bis(o-aminophenoxy)ethane-N,N,N’N’-tetraacetic acid acetoxymethyl ester (BAPTA-AM) were purchased from Sigma-Aldrich(St. Louis, MO). Rhod-2-AM, Fluo-3-AM, chloromethyl-H2DCF-DA (CM-H2DCF-DA), MitoTracker-Red,MitoSOX-Red, 4’6-diamidino-2-phenylindole (DAPI), calcein- acetoxymethyl ester (calcein-AM), and ethidium homodimer-1 (EthD-1) were from Molecular Probes(Carlsbad, CA). 2-aminoethosxydiphenyl borate (2-APB) were obtained from Calbiochem(San Diego, CA). Dantrolene was obtained from Alexis Biochemicals(San Diego, CA). Caspases inhibitors benzyloxy-carbonyl-Val-Ala-Asp-(OMe) fluoromethyl ketone (z-VAD-fmk) was from R&D systems (Minneapolis, MN). The following antibodies were used: anti-β-actin (Abcam); anti-ubiqitin, ATF4 (Santa Cruz Biotechnologies, Santa Cruz, CA); anti-caspase-8, caspase-3 and PDI (Stressgen, BC, Canada); anti-caspase-9 (Novus Biologicals); anti-CHOP, phosphor-eIF2α, total eIF2α, AIP-1/Alix, and LC3B (Cell Signaling,Beverly, MA); anti-c-FLIP (NF6) (Alexis, San Diego, CA); anti-cleaved PARP (Epitomics Inc.); anti-p62 (BD biosciences pharmingen); anti-COX II (Invitrogen); HRP-conjugated anti-rabbit IgG and HRP-conjugated anti-mouse IgG
7
(Molecular Probes).
B. Cell culture of various cancer cell lines
The MDA-MB 435S, MDA-MB 231 and MCF-7 human breast cancer cells were purchased from American Type Culture Collection(ATCC, Manassas, VA). These various cancer cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics (GIBCO-BRL, Grand Island, NY). Cells were maintained in a humidified atmosphere containing 5% CO2at 37℃. CDDO-ME (>98% purity, Cayman) and CDDO-ME (>98% purity, Cayman)dissolved in dimethyl sulfoxide (DMSO) at a concentration of 50 mM and stored at -20℃. These stock solutions were diluted to the required concentration when need.
C. Measurement of cell viability
Cell viability was assessed by double labeling of cells with 2 μM calcein-AM and 4 μM EthD-1. The calcein-positive live cells and EthD-1-positive dead cells were visualized using a fluorescence microscope (Axiovert 200M using Axiovision Release 4.4 and Axiocam HRM digital camera; Zeiss, Oberkohen, Germany) and counted.
8
Cells were washed in PBS and lysed in boiling sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (6.25 mM Tris [pH 6.8], 1% SDS, 10% glycerol, and 5% β-mercaptoethanol). The lysates were boiled for 5 min, separated by SDS-PAGE, and transferred to an Immobilion membrane (Millipore, Bredford, MA, USA). After blocking nonspecific binding sites for 1 h using 5% skim milk, membrane were incubated for 2 h with specific Antibodies. Membranes were then washed three times with TBST and incubated further for 1 h with horseradish peroxidase-conjugated anti-rabbit, -mouse antibody. Visualization of protein bands was accomplished using ECL (Advansta).
E. Immunocytochemistry
MDA-MB 435S breast cancer cellswere plated on chamber slides before drug exposure. After treatments, cells were fixed with acetone/methanol (1:1) for 5 min at -20℃ and blocking in 5% BSA in PBS for 30 min. Fixed cells were incubated overnight at 4℃ with primary antibody and then washed three times in PBS and incubated for 1 h at room temperature with anti-rabbit or anti-mouse Alexa Fluor 488 or 594 (1:500, Molecular Probes). Slides were mounted with ProLong Gold antifade mounting reagent (Molecular probes) and cell staining was visualized with a fluorescence microscope using Zeiss filter sets #46 and #64HE (excitation band pass, 598/25 nm; emission band pass, 647/70 nm).
9
F. Establishment of the stable cell lines in the fluorescence specifically
mitochondria or endoplasmic reticulum
To establish the stable cell lines expressing the fluorescence specifically in mitochondria or the ER, MDA-MB 435S cells were transfected with the pEYFP-Mito of pEYFP-ER vector (Clontech Laboratories, Mountain View, CA). Stable cell lines over-expressing pEYFP-Mito of pEYFP-ER (YFP-Mito of YFP-ER) were selected with the complete media containing 500 μg/mL G418 (Calbiochem, San Diego, CA). Images of mitochondria or ER were obtained from the fluorescence microscopy using a Zeiss filter set #10.
G. Measurement of ROS and mitochondrial superoxide anion
Cells were exposed to 1.5μM CDDO-ME for indicated duration. The cells were stained with 5 μM CM-H2DCF-DAfor 30 min or 2.5 μM MitoSOX red for 20 min at 37℃ in the dark. After being washed with PBS or HBSS with Ca²⁺ and Mg²⁺, and then observed under a fluorescence microscope or flow cytometry using CellQuest software (Becton Dickinson, San Jose, CA).
10
To measure cytosolic Ca²⁺ levels,treated cells were incubated with 2.5μM Fluo-3-AM at 37℃ for 20 min, washed with HBSS, and analyzed immediately by flow cytometry. To measure mitochondrial Ca²⁺ levels, treated cells were incubated with 2.5μM Rhod-2-AM at 4℃ for 30 min, washed with HBSS, further incubated with HBSS at 37℃ for 20 min, and then analyzed by flow cytometry.
I. shRNA-mediated knockdown of proteins
Knockdown of ATG5, Beclin-1, LAMP2 protein in breast cancer cells was achieved by lentiviral infections of viral vectors that express different shRNA directed to ATG5, Beclin-1, LAMP2 mRNA and were purchased from Sigma-Aldrich (ShRNA MISSION). Viruses were generated by transfection of 293T cells with MISSION shRNA vectors and DNRF vector encoding for gag- pol, and CMV-VSVG encoding for envelop glycoprotein of vesicular stomatitis virus. The medium of transfected 293T cells containing lentiviruses was used to infect myoblasts that were further selected with puromycin (3 mg/ml). Knockdown efficiency was analyzed by western blotting. Viral particles that caused maximal repression of ATG5, Beclin-1, LAMP2 mRNA expression relative to control particles were chosen for the knockdown experiments.
J. Transmission electron microscopy
11
2 mM calcium chloride, 0.1 M cacodylate buffer, pH 7.4) for 2 h and washed with cacodylate buffer. Post-fixing was carried out in 1% osmium tetroxide and 1.5% potassium ferrocyanide for 1 h. After dehydration with 50-100% alcohol, the cells were embedded in Poly/Bed 812 resin (Pelco, Redding, CA, USA), polymerized, and observed under electron microscope (EM 902A, Zeiss, Oberkohen, Germany).
K. Reverse transcription–PCR analysis
Total RNA was extracted from MDA-MB 435S cells using the TRIzol reagent (Invitrogen). Reverse transcription-PCR (RT-PCR) was done, following the manufacturer's protocol (TaKaRa Shuzo Co., Otsu, Shiga, Japan). The cDNAs were amplified by PCR (30 s at 94℃, 30 s at 60℃, and 1 min 74℃) with Taq DNA polymerase. Conditions for final analysis were chosen when amplification of mRNA was in the middle of the exponential amplification phase for 1.5 µM CDDO-ME tested. For c-FLIPL, the sense primer 5'-CGGACTATAGAGTGCTGATGG-3' and the antisense primer 5'-GATTATCAGGCAGATTCCTAG-3' (corresponding to a 655-bp region of c-FLIPL) and for glyceraldehyde 3-phosphate dehydrogenase (GAPDH), the sense primer
5′-CGGCCATCACGCCCACAGTTT-3′ and the antisense primer
5′-CGGCCATCACGCCCACAGTTT-3′ (corresponding to a 310 bp region of GAPDH)were used. The PCR cycling conditions (30 cycles) chosen were as follows: (a)
12
30 s at 94℃; (b) 30 s at 56℃ for c-FLIPL and 30 s at 60℃ for GAPDH; (c) 1 min 30 s at 72℃, with a subsequent 10 min extension at 72℃. Reaction products were analyzed on 2% agarose gels. The bands were visualized by ethidium bromide.
L. Statistical analysis
All data were presented as mean ± S.D. (standard deviation) from at least three separate experiments. Student’s t test was applied to evaluate the differences between treated and control groups with cell viability. Data from multiple groups were analyzed by one-way ANOVA, followed by Bonferroni multiple comparison test. For all the tests, the level of significance was values of P < 0.05.
13
III. RESULTS
1.CDDO-ME demonstrates a potent anti-cancer effect on breast cancer
cells.
Triterpenoids, including CDDO and CDDO-ME(Figure 1), the methylester of CDDO, are shown to demonstrate anti-cancer effects on various cancer cells, including lung, prostate and pancreatic cancer cells (Ryu K et al. 2010; Samudio I et al. 2008; Liu Y et
al. 2012; Zou W et al. 2008). In the present study, we examined the cytotoxic effects of
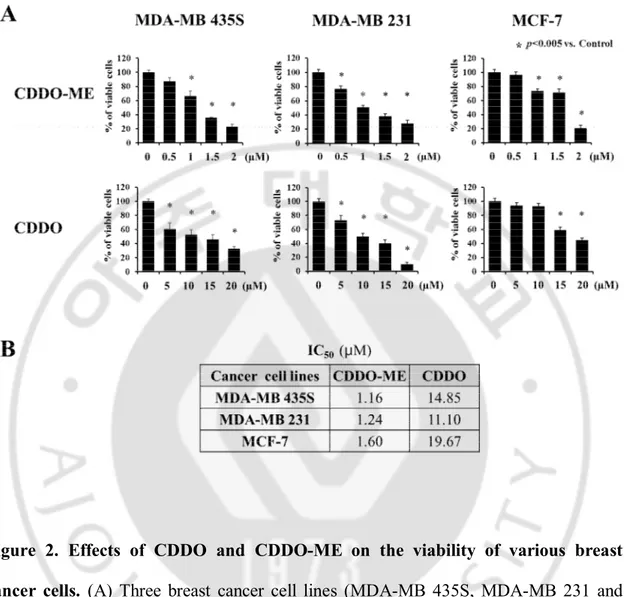
CDDO and CDDO-ME on breast cancer cells. Cell viability assay using calcein-AM and EthD-1 to detect live and dead cells, respectively, were performed following treatment of MDA-MB 435S, MDA-MB 231 and MCF-7 cells with various doses of CDDO and CDDO-ME for 24 h, and IC50s of CDDO and CDDO-ME were assessed. Although both CDDO and CDDO-ME dose-dependently reduced the viabilities of these breast cancer cells, CDDO required about 9 ~ 13 fold higher concentrations to exert a similar cytotoxicity of CDDO-ME in these cells (Figure 2A and 2B). Colony forming assay also showed that the inhibiting effect of CDDO-ME on long-term survival of MDA-MB 435S cells was much stronger than that of CDDO (Figure 3A and 3B). Taken together, these results indicate that CDDO-ME demonstrates a much stronger anti-cancer effect than CDDO on breast anti-cancer cells.
14
15
Figure 2. Effects of CDDO and CDDO-ME on the viability of various breast cancer cells. (A) Three breast cancer cell lines (MDA-MB 435S, MDA-MB 231 and MCF-7) were treated with CDDO or CDDO-ME at the indicated concentrations for 24 h. Cellular viability was assessed using calcein-AM and EthD-1 to detect live and dead cells, respectively. (B) The values of IC50 are the concentration of each drug that is required for 50% inhibition of cell survival in vitro.
16
Figure 3. Dose-dependent effects of CDDO-ME and CDDO on the long-term survival of MDA-MB 435S cells.MDA-MB 435S cells seeded on 12 well-plates were treated with CDDO-ME or CDDO at the indicated concentrations for 12 h and then media were replaced with drug-free media. Following the subsequent incubation for 9 days, cells were stained with 0.5% crystal violet. (A) Representative dishes after clonogenic assay are shown. (B) Colony-forming units were enumerated and expressed as the percentages of control cells.
17
2. CDDO-ME induces paraptosis-like cellular vacuolation prior to
morphologies features of apoptosis in breast cancer cells
To understand the underlying mechanism of the potent cytotoxic effect of CDDO-ME on breast cancer cells, we first examined changes in the cellular morphologies of breast cancer cells following CDDO-ME treatment. Interestingly, we found that severe cellular vacuolation preceded the apoptotic morphologies, including cell shrinkage, cytoplasmic blebbing, and apoptotic bodies, commonly in MDA-MB 435S, MDA-MB 231 and MCF-7 cells (Figure 4). Since a double-membrane vesicle known as autophagosome were observed in cytoplasm during autophagy process, we next examined whether CDDO-ME-induced vacuolation and cell death was associated with autophagy. When we examined the knockdown effects of ATG5, Beclin-1, or LAMP2 on CDDO-ME-induced cellular responses, neither CDDO-ME-CDDO-ME-induced cellular vacuolation, which was observed at 12 h of CDDO-ME post-treatment, nor cell death, which was measured at 24 h of post-treatment, was affected by their knockdown (Figure 5A-5C). In addition, pretreatment with autophagy inhibitors, including 3-methyladenine (3-MA) and chloroquine (CQ), did not affect CDDO-ME-induced vacuolation and cell death (Figure 6A and 6B).Also, when we observed the expression of LC3, known as a autophagy marker, and p62, substrate proteins of autophagy (Johansen T et al. 2011), time-course experiment showed that the protein levels of both LC3B and p62 were significantly increased following CDDO-ME treatment (Figure 7). If CDDO-ME inducedautophagy,
18
we could observe LC3 conversion from LC3-I to LC3-II and p62 degradation (Rogov V and Dötsch Vet al., 2014). However, we just observed accumulation of both LC3B and p62,suggesttingthat autophagy may not be associated with CDDO-ME-mediated cellular responses in these cells.
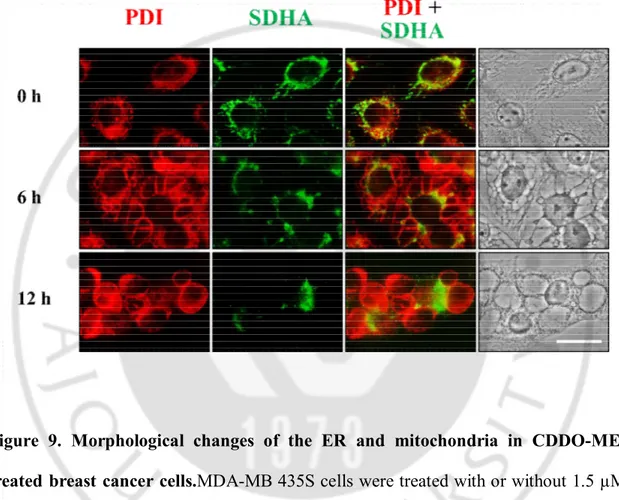
Next, we examined whether CDDO-ME-induced vacuoles were originated from the endoplasmic reticulum and/or mitochondria. For this purpose, we employed MDA-MB 435S sublines stably expressing fluorescence selectively in the ER (YFP-ER cells) or mitochondria (YFP-Mito cells). While the ER appeared as a reticular structure in untreated YFP-ER cells, ER fluorescence exactly co-localized with numerous vacuoles in YFP-ER cells treated with 1.5 µM CDDO-ME for 6 h. At 12 h of CDDO-ME treatment, the sizes of ER-derived vacuoles were further increased, but their numbers were decreased, possibly suggesting the fusion among these vacuoles (Figure 8). While mitochondria in untreated YFP-Mito cells showed a filamentous and elongated structure, mitochondrial fluorescence in the cells treated with CDDO-ME for 6 h revealed fragmented morphology or co-localized at very small vacuoles around the nuclei, but not at easily discernible vacuoles by the phase contrast microscopy. And then, most of mitochondria appeared to be fragmented in YFP-Mito cells treated with CDDO-ME for 12 h(Figure 8). Immunocytochemistry of protein disulfide-isomerase (PDI), an ER-resident protein, and the subunit A of succinate dehydrogenase (SDHA), a mitochondrial protein, showed that PDI expression of a reticulate structure and elongated SDHA expression were detected in untreated MDA-MB 435Scells (Figure 9),
19
similar to ER and mitochondrial morphologies shown in YFP-ER and YFP-Mito cells (Figure 8). At 6 h of 1.5 µM CDDO-ME treatment, large rings of PDI expression and very small rings of SDHA expression were observed. SDHA-expressing mitochondria-derived vacuoles appeared to be localized near the nuclei, whereas PDI-expressing ER-derived vacuoles were peripheral to the mitochondria-ER-derived vacuoles. At 12 h of CDDO-ME treatment, the sizes of ER-derived vacuoles, but not mitochondria-derived vacuoles, were further increased. These results suggest that CDDO-ME-induced vacuolation is mainly resulted from the dilation of the ER in these breast cancer cells.
Since ER dilation rather than mitochondrial dilation was noted following CDDO-ME treatment, we next examined whether CDDO-CDDO-ME induces ER stress(Ron D et al., 2007; Huber AL et al., 2013; Szegezdi E et al., 2006; Bertolotti et al., 2000; Liu CY et
al., 2003). Western blotting showed that CDDO-ME treatment markedly accumulated
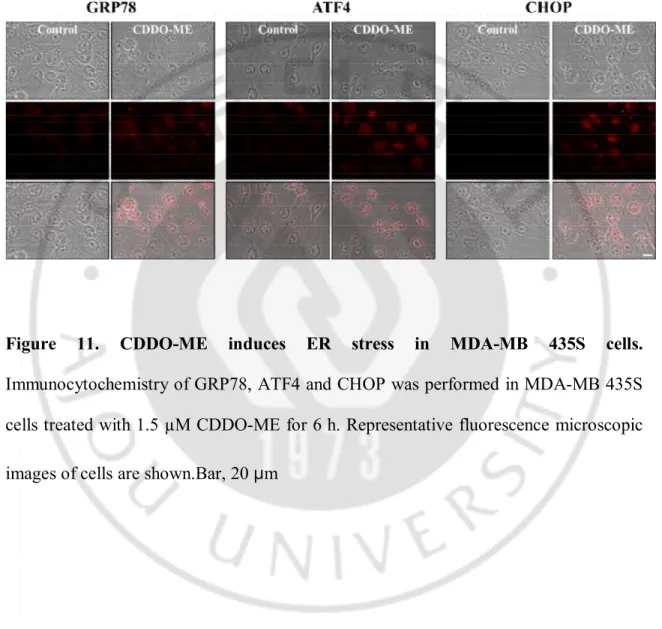
the protein levels of GRP78, phosphorylated eIF2α, ATF4, and CHOP (Figure 10).Interestingly, we found that caspase-4 was also cleaved from 12 h to 24 h by CDDO-ME treatment.We confirmed again that CDDO-ME induces ER stress by immunocytochemistry. When we performed immunocytochemistry of GRP78, ATF4 or CHOP, we observed that the expression levels increased in the nucleus (Figure 11). These results indicated that CDDO-ME is an effective ER stress inducer in breast cancer cells.
To observe the changes in cellular organelles in more detail following CDDO-ME treatment, we performed electron microscopy. Untreated MDA-MB 435S cells
20
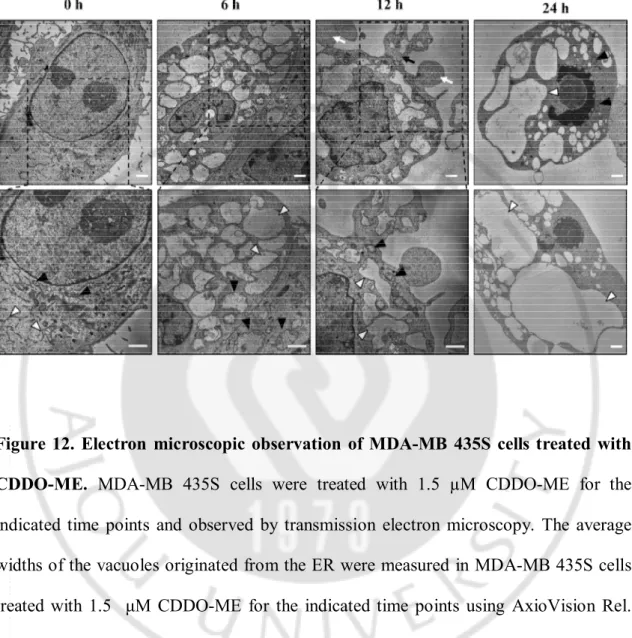
possessed the ER structures with elongated sacs surrounded by one-layer membrane and mitochondria with intact cristae (Figure 12). In cells treated with CDDO-ME for 6 h, cellular space was mostly occupied with expanded structures of the ER and the fusion among the ER was further progressed after 12 h of treatment. When we quantitatively measured the sizes of the ER-derived vacuoles, their average width were 2.468 µm, 2.677 µm and 4.405 µm at 6, 12, and 24 h of CDDO-ME treatment, respectively. In contrast, swollen mitochondria were frequently observed at 6 h of treatment, but their sizes were rather reduced at 12 h. These results suggest that cells underwent a transient mitochondrial swelling in response to CDDO-ME treatment. Or mitochondria might undergo a fusion at 6 h of CDDO-ME treatment and then fission at 12 h. Very interestingly, the dilation of ER was accompanied by cytoplasmic blebbing and formation of apoptotic bodies at 12 h of post-treatment. At 24 h, chromatin condensation and DNA fragmentation were detected (Figure 12).
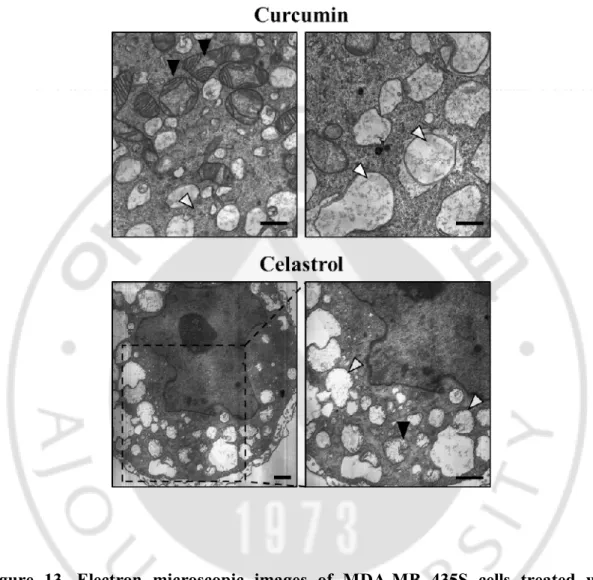
Interestingly, we observed that vacuolation induced by CDDO-ME is similar to vacuolation by paraptosis inducers, curcumin or celatrol(Yoon MJ et al., 2010; Yoon MJ and Lee AR et al., 2014). Electron microscopic images of breast cancer cells treated with CDDO-ME, curcumin or celastrol showed that all of these drugs induced dilation of the ER.Collectively, these results indicate that CDDO-ME induces paraptosis-like morphologies and subsequently apoptotic ones in these breast cancer cells (Figure 13).
Therefore, we next investigated whether paraptosis is also involved in CDDO-ME-induced cell death. Although the underlying mechanisms of paraptosis are not clearly
21
understood, paraptosis is known to be inhibited by cycloheximide, because paraptosis required new gene transcription and translation(Sperandio Set al. 2004). When we tested whether CDDO-ME-induced cell death is affected by cycloheximide pretreatment, neither cell death nor ER-derived vacuolation induced by CDDO-ME was inhibited by cycloheximide pretreatment (Figure 14A and 14B). However, cells treated with curcumin and celastrol, inducing paraptosis, were completely blocked by cycloheximide. In YFP-ER cells, although we pretreated with cycloheximide and further treated 1.5 µM CDDO-ME for 6 h, ER fluorescence still co-localized with numerous vacuoles. These results also suggested that CHX didn’t block ER dilation induced by CDDO-ME (Figure 15).
Chatellard-Causse C et al. reported that Alix interaction with endophilins can induce intracellular vacuole formation (Chatellard-Causse C et al. 2002). Alix was determined to be "the first specific inhibitor" of paraptosisby us and other groups (S. Sperandioet al. 2004; F. Valamaneshet al. 2007). Since Alix was reported a paraptosis inhibitor, we examined the changes in Alix protein levels following CDDO-ME treatment. We found that expression of Alix was not altered by CDDO-ME treatment, whereas it was markedly reduced by curcumin or celastrol, paraptosis inducers (Figure 16), indicating that CDDO-ME didn’t induce paraptosis.
Paraptosis is known to lack caspase activation and is insensitive to caspase inhibitors (Gao X et al. 2011; Kim Y et al. 2002). When we examined the expression of caspases, treatment of MDA-MB 435S cells with 1.5 µM CDDO-ME induced the proteolytic
22
processing of caspase-8 from 12 h and caspase-9 and -3 from 18 h. PARP, a substrate of caspase-3, was also cleaved from 18 h of CDDO-ME post-treatment (Figure 17). Furthermore, pretreatment with z-VAD-fmk significantly and commonly inhibited CDDO-ME-induced cell death in MDA-MB 435S, MDA-MB 231, and MCF-7 cells (Figure 18). When we examined the location of cytochrome c following CDDO-ME treatment, we found that CDDO-ME treatment released cytochrome c from mitochondria into cytosol(Figure 19A). Cleaved PARP was detected in shrunken MDA-MB 435S cells treated with CDDO-ME for 24 h, but not in all the vacuolated cells treated with CDDO-ME for 24 h (Figure 19B). These results indicate that caspase-3 may be activated in parallel with cellular shrinkage. Finally, we sought to examine whether CDDO-ME-induced cell death was associated with changes in cell cycle distribution.MDA-MB 435S cells were treated with CDDO-ME and subjected to fluorescence activated cell sorting analysis, and the percentages of sub-G1cells were quantified (Figure 20). Treatment with CDDO-ME alter the cell cycle distribution. In cells treated with 1.5 μM CDDO-ME for 48 h, the percentage of sub-G1 cells was markedly increased, suggesting that CDDO-ME induced apoptotic cell death. Taken together, CDDO-ME triggers extensive vacuolation mainly derived from the ER, but ultimately kills breast cancer cells via caspase-mediated apoptosis.
23
Figure 4. CDDO-ME induces extensive dilation prior to apoptotic cell death in MDA-MB 435S cells. Three breast cancer cells were treated with 1.5 µM CDDO-ME for the indicated time points and observed under the phase contrast microscopy.Bars, 20 μm.
24
Figure 5. Knockdown of ATG5, beclin-1, LAMP2 does not block cell death andvacuolationinduced by CDDO-ME. (A) MDA-MB 435S cells were treated with the lentivirus encoding the control non-targeting RNA, ATG5, Beclin-1 or LAMP2 shRNA and further treated with or without 1.5 μM CDDO-ME for 24 h. Knockdown of these gene products was confirmed by Western blots. (B) Cellular viability was assessed using calcein-AM and EthD-1.(C) Cells were observed under a phase contrast microscope. Bar, 20 µm.
25
Figure 6. Effects of autophagy inhibitors on CDDO-ME-induced vacuolation and cell death. (A) MDA-MB 435S cells were pretreated with 3-MA orchloroquine for 30 min and further treated with 1.5 µM CDDO-ME for 24 h. Cell were observed under a phase contrast microscope.Bar, 20 µm. (B)Cellular viability was assessed using calcein-AM and EthD-1.
26
Figure 7. Autophagy-related proteins, LC3B and p62, were accumulated in MDA-MB 435S cells treated with CDDO-ME.MDA-MDA-MB435S cells were treated with 1.5 µM CDDO-ME for indicated time points. Whole cell extracts were prepared and subjected to western blotting using LC3B and p62 antibodies. β-actin was used as a loading control.The fold change of protein levels compared to 0 h was determined by a densitometric analysis.
27
Figure 8. CDDO-ME induces dilation of the ER and mitochondrial fragmentation.YFP-Mito and YFP-ER cells were treated with 1.5 µM CDDO-ME for indicated time points and then observed under a fluorescence microscope. Bar, 20 μm.
28
Figure 9. Morphological changes of the ER and mitochondria in CDDO-ME-treated breast cancer cells.MDA-MB 435S cells were CDDO-ME-treated with or without 1.5 µM CDDO-ME for 12 h. Immunocytochemistry using anti-SDHA (green) and anti-PDI (red) antibodies was performed and the representative phase contrast and fluorescence microscopic images of cells are shown.Bar, 20 µm.
29
Figure 10. CDDO-ME increases the protein levels of ER stress-associated proteins in MDA-MB 435S cells. Cells were treated with 1.5 µM CDDO-ME for the indicated time points or indicated doses of CDDO-ME for 24 h and then Western blotting was performed. β-actin was used as a loading control in Western blots. The fold change of protein levels compared to β-actin was determined by a densitometric analysis.
30
Figure 11. CDDO-ME induces ER stress in MDA-MB 435S cells. Immunocytochemistry of GRP78, ATF4 and CHOP was performed in MDA-MB 435S cells treated with 1.5 µM CDDO-ME for 6 h. Representative fluorescence microscopic images of cells are shown.Bar, 20 µm
31
Figure 12. Electron microscopic observation of MDA-MB 435S cells treated with CDDO-ME. MDA-MB 435S cells were treated with 1.5 µM CDDO-ME for the indicated time points and observed by transmission electron microscopy. The average widths of the vacuoles originated from the ER were measured in MDA-MB 435S cells treated with 1.5 μM CDDO-ME for the indicated time points using AxioVision Rel. 4.8 software (Zeiss). White arrow heads, ER; Black arrow heads, mitochondria; White arrows, apoptotic bodies; Black arrows, blebbing. Bars, 2 µm.
32
Figure 13. Electron microscopic images of MDA-MB 435S cells treated with curcumin or celastrol. MDA-MB 435S cells were treated with 40 µM curcumin for 24 h or 2 µM celastrol for 12 h and observed by transmission electron microscopy; White arrow heads, ER; Black arrow heads, mitochondria. Bars, 2 µm.
33
Figure 14. Cycloheximide, a translation inhibitor, does not block CDDO-ME-induced cell death and vacuolation. (A) MDA-MB 435S cells were pretreated with the indicated concentrations of CHX for 30 min and further treated with 1.5 µM CDDO-ME for 24 h. Cellular viability was assessed using calcein-AM and EthD-1. (B) Cells were observed under a phase contrast microscope. Bar, 20 µm
34
Figure 15. CDDO-ME-induced ER dilation is not blocked by cycloheximide.YFP-ER cells pretreated with CHX and further treated with or without 1.5 μM CDDO-ME for 6 h were stained with Mitotracker Red and then observed using phase contrast and fluorescence microscopy. Bar, 20 µm
35
Figure 16. Change in protein expression of Alix.MDA-MB 435S cells were treated with 1.5 µM CDDO-ME for the indicated time points or 40µM curcumin or 2µM celastrol for 24 h and then Western blotting was performed. β-actin was used as a loading control in Western blots. The fold change of protein levels compared to β-actin was determined by a densitometric analysis.
36
Figure 17. Activation of caspases in MDA-MB 435S cells treated with CDDO-ME.MDA-MB 435S cells were treated with indicated doses of CDDO-ME for 24 h. Whole cell extracts were prepared from the treated cells and subjected to Western blotting. β-actin was used as a loading control in Western blots. The fold change of protein levels compared to β-actin was determined by a densitometric analysis.
37
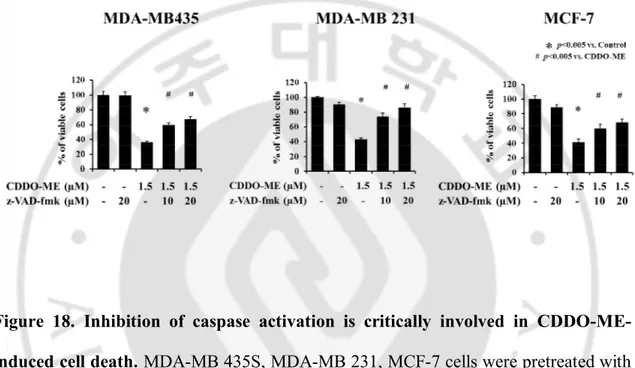
Figure 18. Inhibition of caspase activation is critically involved in CDDO-ME-induced cell death. MDA-MB 435S, MDA-MB 231, MCF-7 cells were pretreated with the indicated concentrations of z-VAD-fmk for 30 min and further treated with 1.5 µM CDDO-ME for 24 h. Cellular viability was assessed using calcein-AM and EthD-1.
38
Figure 19. CDDO-ME induces chromatin condensation and release of cytochrome c. (A) DAPI staining were performed in MDA-MB 435S cells untreated or treated with 1.5 µM CDDO-ME for 24 h. Representative fluorescence microscopic images of cells are shown. (B) Immunocytochemistry of the cytochrome c and the subunit I of cytochrome c oxidase (COX IV) was performed in MDA-MB 435S cells treated with 1.5 µM CDDO-ME for 24 h, as described in Materials and Methods. Representative fluorescence microscopic images of cells are shown. Bars, 20 μm.
39
Figure 20. Effect of CDDO-ME on MDA-MB 435S cell cycle progression.MDA-MB 435S cells were treated with or without 1.5 µM CDDO-ME for 48 h. Cells were stained with propidium iodide and FACS analysis.
40
3. Ca
2+influx is crucial for CDDO-ME-induced vacuolation and
subsequent apoptotic cell death
Since CDDO-ME induces severe ER-derived vacuolization and ER is a major reservoir of intracellular Ca2+ in the cells, we next tested whether CDDO-ME induces the perturbation of intracellular Ca2+ homeostasis. Both fluorescence microscopy and flow cytometry using Fluo-3 (a cell-permeable Ca2+-indicator dye) demonstrated that treatment of MDA-MB 435S cells with 1.5 µM CDDO-ME dramatically increased the intracellular Ca2+ levels, which peaked at 4 h post-treatment (Figure 21). We further tested whether CDDO-ME increased mitochondrial Ca2+ levels by flow cytometry using Rhod-2 (an indicator dye for mitochondrial Ca2+). CDDO-ME treatment also increased the mitochondrial Ca2+ levels with a peak at 4 h post-treatment, but the extent of the increase in Rhod-2 staining was much less than that of Fluo-3 (Figure 22).
Next, we investigated the sources of the increased Ca2+ levels in response to CDDO-ME. Intracellular Ca2+ levels can be increased either by influx from the extracellular milieu or by the release from the ER, via two major signaling pathways, IP3 receptor (IP3R) and ryanodine receptor (RyR) (Marks,A.R.et al. 1992). We found that two scavengers of extracellular Ca2+, BAPTA and EGTA, dose-dependently inhibited the cell death in MDA-MB 435S cells. Furthermore, BAPTA plus BAPTA-AM or EGTA plus BAPTA-AM demonstrated a further cytoprotective effect on CDDO-ME-induced cell death, compared to a single treatment(Figure 23). In contrast, pretreatment with
41
neither dantrolene, a specific inhibitor of the RyR nor 2-APB, a specific inhibitor of IP3R, did affect CDDO-ME-induced cell death. These results suggest that the influx of extracellular Ca2+ may critically contribute to this cell death, rather than Ca2+ release from the ER. Furthermore, we examined whether the increase in Ca2+ levels has a functional significance in CDDO-ME-induced cell death. We found that scavenging of intracellular Ca2+ employing BAPTA-AM dose-dependently inhibited the death in MDA-MB 435S cells treated with 1.5 µM CDDO-ME (Figure 23), suggesting that the increase in Ca2+ in the cells may be critically associated with CDDO-ME-induced cell death.
We next asked whether scavenging of intra- and/or extracellular Ca2+ affected CDDO-ME-induced dilation of the ER using YFP-ER and YFP-mito cells. We found that scavenging of intracellular Ca2+ using BAPTA-AM or scavenging of extracellular Ca2+ using BAPTA or EGTA significantly inhibited CDDO-ME-induced ER dilation in MDA-MB 435S cells treated with 1.5 µM CDDO-ME.When we pretreated BAPTA-AM plus BAPTA or BAPTA-BAPTA-AM plus EGTA further inhibited it, compared with a single treatment (Figure 24A and 24B). These Ca2+ inhibitors had no effect on mitochondrial fragmentation (Figure 25A and 25B). Also, BAPTA plus BAPTA-AM markedly inhibited PARP cleavage caused by CDDO-ME treatment (Figure 26). These results suggest that Ca2+ influx and a resultant increase in Ca2+ levels may critically contribute to ER dilation, promoting apoptotic cell death.
Increase in Ca2+ levels by CDDO-ME was also observed in MDA-MB 231 and MCF-7 cells (Figure 2MCF-7). In addition, pretreatment with either BAPTA or BAPTA-AM
42
significantly attenuated CDDO-ME-induced cell death also in these breast cancer cells (Figure 28). Taken together, these results indicate that Ca2+ influx may be a common and critical signal for CDDO-ME-induced vacuolation and subsequent cell death in breast cancer cells.
43
Figure 21. CDDO-ME increases intracellular Ca2+ levels. MDA-MB 435S cells
treated with 1.5 µM CDDO-ME for the indicated time points were stained with 2.5 µM Fluo-3 and observed under the phase contrast and fluorescence microscope or processed for FACS analysis.Fluo-3 fluorescence intensities (FI) in cells treated with 1.5 µM CDDO-ME were compared with that of untreated cells and denoted in the graph (left). Histogram for the cells treated with 1.5 µM CDDO-ME for 4 h is shown (right). X axis, fluorescence intensity, Y axis, relative number of cells. Bar, 20 µm.
44
Figure 22. CDDO-ME does not affect mitochondrial Ca2+levels. MDA-MB 435S cells treated with 1.5 µM CDDO-ME for the indicated time points were stained with 2.5 µM Rhod-2 and processed for FACS analysis. Rhod-2 fluorescence intensities (FI) in cells treated with 1.5 µM CDDO-ME were compared with that of untreated cells and denoted in the graph (left). Histogram for the cells treated with 1.5 µM CDDO-ME for 4 h is shown (right). X axis, fluorescence intensity, Y axis, relative number of cells.
45
Figure 23. Effect of Several Ca2+ inhibitors on MDA-MB 435S cells treated with CDDO-ME. MDA-MB 435S cells were untreated or pretreated with the indicated Ca2+ antagonists (BAPTA-AM, BAPTA, EGTA, dantrolene, 2-APB) at the indicated concentrations for 30 min and further treated with 1.5 µM CDDO-ME for 24 h. Cellular viability was assessed using calcein-AM and EthD-1.
46
Figure 24. Dilation of the ER is blocked by BAPTA, BAPTA-AM and EGTA.YFP-ER cells were untreated or pretreated with the indicated specific inhibitors and further treated with 1.5 µM CDDO-ME for 6 h. Cells were observed under a fluorescence microscope. Bars, 20 µm. The changes in the widths of the ER-derived vacuoles were quantitatively measured using AxioVision Rel. 4.8 software. Results were repeated in three other experiments. In each experiment, 50 cells were scored as described in Materials and Methods section. Bar, 20 µm
47
Figure 25. Mitochondrial fragmentation is not associated with Ca2+ influx.YFP-Mito cells were untreated or pretreated with the indicated specific inhibitors and further treated with 1.5 µM CDDO-ME for 6 h. Cells were observed under a fluorescence microscope. Bars, 20 µm. The changes in the widths of mitochondria-derived vacuoles were quantitatively measured using AxioVision Rel. 4.8 software. Results were repeated in three other experiments. In each experiment, 50 cells were scored as described in Materials and Methods section. Bar, 20 µm
48
Figure 26. PARP cleavage is blocked by BAPTA-AM plus BAPTA in CDDO-ME-treated cells.MDA-MB 435S cells were preCDDO-ME-treated with 5 µM BAPTA-AM plus 5 µM BAPTA, and further treated with 1.5 µM CDDO-ME for 24 h followed by Western blotting of cleaved PARP. β-actin was used as a loading control in Western blots and The fold change of protein levels compared to β-actin was determined by a densitometric analysis.
49
Figure 27. CDDO-ME increases intracellular Ca2+levels in other breast cancer cells.MDA-MB 231 or MCF-7 cells treated with 1.5 µM CDDO-ME for 3 h, 6 h, respectively, were stained with 2.5 µM Fluo-3 and observed under the phase contrast and fluorescence microscope or processed for FACS analysis.
50
Figure 28. BAPTA plus BAPTA-AM block CDDO-ME-induced cell death in other breast cancer cells. Cells were untreated or pretreated with the indicated Ca2+ antagonists at the indicated concentrations for 30 min and further treated with 1.5 µM CDDO-ME for 24 h. Cellular viability was assessed using calcein-AM and EthD-1.
51
4. Cross-modulation between Ca
2+influx and ROS generation critically
contributes to CDDO-ME-induced vacuolation and subsequent
apoptosis
Previously, ROS were reported to be critically involved in the growth inhibition and apoptosis induced by CDDO-ME in various cancer cells (Deeb D et al. 2010; Gao X et
al. 2011; Deeb D et al. 2012). Thus, we examined whether CDDO-ME generates ROS,
if so, whether this contributes to the vacuolation and subsequent apoptosis in breast cancer cells. An assessment of ROS levels using CM-H2DCF-DA showed that 1.5 µM CDDO-ME treatment increased ROS levels, which peaked at 4 h post-treatment (Figure 29). Increase in ROS levels were also observed in MDA-MB-231 and MCF-7 cells treated with 1.5 µM CDDO-ME for 4 h (Figure 29). When we examined the functional significance of ROS generation in CDDO-ME-induced cell death, pretreatment with either N-acetyl cysteine (NAC) or glutathione (GSH) significantly blocked the cell death in these three breast cancer cell lines (Figure 30).
An analysis of mitochondrial superoxide levels using MitoSOX-Red showed that treatment of MDA-MB 435S cells with CDDO-ME did not significantly increase mitochondrial superoxide levels and pretreatment with MnTBAP, the MnSOD mimetic, did not inhibit this cell death (Figure 31), suggesting that generation of mitochondrial superoxide may not be critically involved in CDDO-ME-induced cell death.
Next, we examined whether ROS generation is required for CDDO-ME-induced ER dilation. We found that NAC or GSH pretreatment very effectively blocked
CDDO-ME-52
induced dilation of the ER in YFP-ER cells. Similar to Ca2+ influx, ROS generation also had no effect on mitochondrial fragmentation (Figure 32). Taken together, ROS critically contributes to ER-derived vacuolation and subsequent cell death.
Since both intracellular Ca2+ and ROS levels were increased with a peak following CDDO-ME treatment and they both were important for vacuolation and subsequent cell death, we next asked the relationship between Ca2+ and ROS in this process. Flow cytometry using Fluo-3 showed that CDDO-ME-induced increase in Ca2+ levels were effectively blocked not only by BAPTA plus BAPTA-AM but also by NAC pretreatment (Figure 33A). In addition, CM-H2DCF-DA staining showed that CDDO-ME-induced ROS generation was effectively blocked not only by NAC but also BAPTA plus BAPTA-AM (Figure 33B). Therefore, these results indicate that there exists the cross-modulation between Ca2+ and ROS during in the progression of CDDO-ME-induced ER-derived vacuolation and subsequent apoptosis.
53
Figure 29. CDDO-ME induces ROS generation in breast cancer cells.MDA-MB 435S, MDA-MB 231, MCF-7 cells were treated with 1.5 µM CDDO-ME for the indicated time points, exposed to 5 µM CM-H2DCF-DA for 30 min and analyzed by flow cytometry. CM-H2DCF-DA fluorescence intensities (FI) were assessed and total ROS levels were compared between cells treated with and without CDDO-ME for the indicated durations. The fold changes of CM-H2DCF-DA FI are shown in the graph (left). Histogram for the cells treated with 1.5 µM CDDO-ME for 4 h is shown (right). X axis, fluorescence intensity, Y axis, relative number of cells.
54
Figure 30. Antioxidants block CDDO-ME-induced cell death. Cells were pretreated with the indicated concentrations of antioxidants (NAC and GSH) for 30 min and further treated with 1.5 µM CDDO-ME for 24 h. Cellular viability was assessed using calcein-AM and EthD-1.
55
Figure 31. CDDO-ME treatment had no effect on mitochondrial ROS generation.(A) MDA-MB 435S cells were treated with 1.5 µM CDDO-ME for the indicated time points, exposed to 5 µM MitoSox-Red for 20 min and analyzed by flow cytometry. (B) Cells were pretreated with the indicated concentrations of MnTBAP for 30 min and further treated with 1.5 µM CDDO-ME for 24 h. Cellular viability was assessed using calcein-AM and EthD-1.
56
Figure 32. Antioxidants blocked dilation of the ER induced by CDDO-ME.YFP-ER and YFP-mito cells were pretreated with NAC or GSH and further treated with 1.5 µM CDDO-ME for 6 h, and then observed under the fluorescence and phase contrast microscope. The changes in the widths of mitochondria-derived vacuoles and the ER-derived vacuoles were quantitatively measured using AxioVision Rel. 4.8 software. Results were repeated in three other experiments. In each experiment, 50 cells were scored as described in Materials and Methods section.
57
Figure 33. Cross-modulation between Ca2+ and ROS generation. (A) MDA-MB 435S cells were pretreated with BAPTA-AM+BAPTA or NAC and further treated with or without 1.5 µM CDDO-ME for 4 h, exposed to 2.5 µM Fluo-3 for 20 min and analyzed by flow cytometry. (B) MDA-MB 435S cells were pretreated with BAPTA-AM+BAPTA or NAC and further treated with or without 1.5 µM CDDO-ME for 4 h, exposed to 5 µM CM-H2DCF-DA and for 30 min and analyzed by flow cytometry.
58
5. c-FLIP
Ldownregulation plays a critical role in CDDO-ME-induced
apoptotic cell death, but not in vacuolation
To clarify the possible contributing factor(s) that lead to caspase-dependent apoptosis by CDDO-ME, we tested whether CDDO-ME modulates the expression of caspase antagonists. Among tested IAPs, protein levels of c-IAP2 were not altered in MDA-MB 435S cells by 1.5 µM CDDO-ME treatment, although those of XIAP and c-IAP1 protein levels were slightly reduced from 12 or 24 h of CDDO-ME post-treatment (Figure 34). In contrast, the protein levels of c-FLIPL, the caspase-8 inhibitor (Kataoka T et al. 2005; Safa AR. et al. 2012), were rapidly and strikingly reduced from 3 h of CDDO-ME treatment, although c-FLIPS protein levels were not detected in the absence or presence of CDDO-ME. In addition, CDDO-ME treatment commonly and dose-dependently reduced the protein levels of c-FLIPL in MDA-MB 435S, MDA-MB 231 and MCF-7 cells (Figure 35).
When we analyzed whether CDDO-ME affected the protein stability of c-FLIPL by blocking of protein synthesis, c-FLIPL protein levels were reduced from 3 h of cycloheximide (Figure 36A). In contrast, those of c-FLIPL were decreased from 1 h by treatment with cycloheximide plus CDDO-ME, indicating that CDDO-ME treatment reduces the protein stability of c-FLIPL. In addition, we found that CDDO-ME progressively dowregulated c-FLIPL mRNA levels by RT-PCR analysis (Figure 36B).
59
Taken together, these results suggest that CDDO-ME potently downregulates c-FLIPL both at the transcriptional and post-transcriptional control.
Since the expression of c-FLIP was previously reported to be modulated by proteasome activity (Fukazawa Tet al. 2001; Zhong Q et al. 2005), we further tested the effect of the proteasome inhibitors on the protein levels of c-FLIPL. We found that CDDO-ME-induced reduction of c-FLIPL protein levels were effectively restored by pretreatment with either MG132 or bortezomib (Figure 37), suggesting that proteasome-mediated degradation of c-FLIPL protein may be involved in CDDO-ME-induced downregulation.
To investigate the functional significance of c-FLIP downregulation during CDDO-ME-induced apoptosis, we next investigated whether exogenously expressed c-FLIPL could block ME-induced cell death. When we examined the effect of CDDO-ME using MDA-MB 435S sublines overexpressing c-FLIPL, we found that c-FLIPL overexpression significantly attenuated CDDO-ME-induced cell death (Figure 38A and 38B). Interestingly, c-FLIPL overexpression did not affect CDDO-ME-induced vacuolation, but inhibited cellular shrinkage and formation of apoptotic bodies, which were detected in the control MDA-MB 435S sublines treated with CDDO-ME for 24 h (Figure 38C).In addition, c-FLIPL overexpression blocked CDDO-ME-induced cleavage of PARP, similar to the effect of z-VAD-fmk (Figure 38A), suggesting that c-FLIP downregulation may play a critical role in CDDO-ME-mediated apoptotic cell death.Similar to these results, pretreatment with z-VAD-fmk also blocked apoptotic morphologies, which were detected in the control cells treated with CDDO-ME for 24 h,
60
but not vacuolation, which were observed in cells treated with CDDO-ME for 6 h(Figure 39A and 39B).
We further investigated the relationship between Ca2+, ROS and c-FLIP downregulation in the progression of CDDO-ME-induced cell death. We found that pretreatment with neither BAPTA plus BAPTA-AM nor NAC did block induced downregulation of c-FLIP (Figure 40). These results suggest that CDDO-ME-induced c-FLIP downregulation is independent of ER-derived vacuolation that is mainly controlled by the increase in intracellular Ca2+ and ROS. However, CDDO-ME-induced c-FLIPL downregulation may contribute to switch the balance of these breast cancer cells.
61
Figure 34. Change in protein levels of caspase antagonists in MDA-MB 435S cells treated with CDDO-ME. MDA-MB 435S cells were treated with 1.5 µM CDDO-ME for the indicated time points or indicated doses of CDDO-ME for 24 h and then Western blotting was performed. β-actin was used as a loading control in Western blots. The fold change of protein levels compared to β-actin was determined by a densitometric analysis.
62
Figure 35. CDDO-ME downregulates c-FLIPL in breast cancer cells.Cells were
treated with 1.5 µM CDDO-ME for the indicated time points or indicated doses of CDDO-ME for 24 h and then Western blotting was performed. β-actin was used as a loading control in Western blots. The fold change of protein levels compared to β-actin was determined by a densitometric analysis.
63
Figure 36. CDDO-ME modulates c-FLIPL in both transcriptional and
post-transcriptional level.(A) MDA-MB 435S cells were treated with or without 1.5 μM CDDO-ME in the presence of CHX (2 μM) for the indicated time periods. The band intensities of c-FLIPL were measured by a densitometric analysis. (B) The mRNA expression levels of c-FLIPL were determined by RT-PCR. The level of GAPDH was used as loading controls.
64
Figure 37. CDDO-ME downregulates c-FLIPL expression by proteasome system.
Cells were pretreated with 1 µM MG132 or 10 nM bortezomib and further treated with 1.5 µM CDDO-ME for 24 h followed by Western blotting. β-actin was used as a loading control.
66
Figure 38.c-FLIP downregulation is critical for apoptotic cell death but not for vacuolation by CDDO-ME.(A) Vector cells (MDA-MB 435S/Vec) and c-FLIPL overexpressed cells (MDA-MB 435S /c-FLIP) were treated with 1.5 µM CDDO-ME for 24 h. Overexpression of c-FLIP was confirmed by Western blotting using anti-c-FLIPL antibody. β-actin was used as a loading control in Western blots. Cellular viability was assessed using calcein-AM and EthD-1. (B) Cells were observed under the phase contrast microscopy. Bars, 20 µm.
67
Figure 39.Overexpression of c-FLIP has similar effectsto z-VAD-fmk. (A)MDA-MB 435S cells were pretreated with 10 µM z-VAD-fmk and further treated with 1.5 µM CDDO-ME for the indicated time points and observed under the phase contrast microscopy. Bars, 20 µm. (B) MDA-MB 435S cells were pretreated with 10 µM z-VAD-fmk and further treated with 1.5 µM CDDO-ME for 24 h followed by Western blotting. β-actin was used as a loading control in Western blots.
68
Figure 40.c-FLIP downregulation is not associated with increase of both intracellular Ca2+ and ROS levels.MDA-MB 435S cells were pretreated with NAC or BAPTA+BAPTA-AM and further treated with 1.5 µM CDDO-ME for 24 h followed by Western blotting of c-FLIPL. β-actin was used as a loading control in Western blots.
69
6. Higher increase in intracellular Ca
2+and ROS levels as well as
c-FLIP downregulation may contribute to a more potent anti-cancer
effect of CDDO-ME, compared to CDDO
Finally, we investigated whether the difference in the cytotoxicity of CDDO-ME and CDDO in breast cancer cells may be associated with their differential modulation of Ca2+, ROS and c-FLIP. Flow cytometry using Fluo-3 and CM-H2DCF-DA showed that Ca2+ and ROS levels were dose-dependently increased in MDA-MB 435S cells treated with CDDO-ME for 4 h (Figure 41). In contrast, Ca2+ levels, but not ROS levels, were slightly increased by the same treatment with CDDO.
Interestingly, the protein levels of c-FLIPL were rather strikingly accumulated by 20 µM CDDO, whereas they were markedly reduced by CDDO-ME (Figure 42). Western blotting of ubiquitin showed that poly-ubiquitinated proteins were dramatically accumulated by CDDO, in parallel with accumulation of c-FLIPL. These results suggest the possibility that CDDO-mediated proteasomal inhibition may increase the protein levels of c-FLIPL, resulting in inefficient activation of caspases and cleavage of PARP.
Collectively, these results indicate that CDDO-ME and CDDO do not share the molecular targets for their anti-cancer effects. We cannot exclude the possibility that upregulation of c-FLIPL in response to CDDO may hinder breast cancer cells die via apoptosis, an effective death mode.
70
Figure 41.A more potent anti-cancer effect of CDDO-ME than that of CDDO may be due to enhanced Ca2+ influx, ROS generation.MDA-MB 435S cells treated with or without indicated dose of CDDO-ME or CDDO for 4 h were stained with 2.5 µM Fluo-3 or 5 µMCM-H2DCF-DA and processed for FACS analysis.
71
Figure 42.The protein levels of c-FLIPL were accumulated by CDDO.MDA-MB
435S cells were treated with 1.5 µM CDDO-ME or 20 µM CDDO for 24 h followed by Western blotting. β-actin was used as a loading control.
72