Independent Regulation of Endothelial Nitric Oxide Synthase by Src and Protein Kinase A in Mouse Aorta Endothelial Cells
Yong Chool Boo*
Department of Molecular Medicine, Kyungpook National University School of Medicine, Daegu, 700-422, Korea
Received June 23, 2005; Accepted August 24, 2005
Endothelial nitric oxide synthase (eNOS) plays a critical role in vascular biology and pathophysiology.
Its activity is regulated by multiple mechanisms such as calcium/calmodulin, protein-protein interactions, sub-cellular locations and phosphorylation at various sites. Phosphorylation of eNOS- Ser1177 (based on mouse sequence) has been identified as an important mechanism of eNOS activation.
However, signaling pathway leading to it phosphorylation remains controversial. The regulation of eNOS-Ser1177 phosphorylation by Src and protein kinase A (PKA) was investigated in the present study using cultured mouse aorta endothelial cells. Expression of a constitutively active Src mutant in the cells enhanced phosphorylation of eNOS and protein kinase B (Akt). The Src-stimulated phosphorylation was not attenuated by the expression of a dominant negative PKA regulatory subunit.
Neither activation nor inhibition of PKA activity had any significant effect on tyrosine phosphorylation of activation or inactivation site in Src. Based on the results of this study, it is suggested that Src/Akt pathway and PKA signaling may regulate eNOS phosphorylation independently. The existence of multiple mechanisms for eNOS phosphorylation may guarantee endothelial nitric oxide production in various cellular contexts which is essential for maintenance of vascular health.
Key words: nitric oxide, eNOS, Src, PKA, mouse aorta endothelial cells Nitric oxide (NO) produced from endothelium plays critical
roles in vascular biology and pathophysiology.1) Endothelium constitutively expresses endothelial nitric oxide synthase (eNOS) which catalyzes NO production from L-arginine and oxygen.2) The essential role of eNOS in the regulation of vascular tone has been supported by eNOS knockout mice which have elevated systemic blood pressures relative to control littermates.3, 4)
eNOS is a distinct Ca2+/Calmodulin (CaM)-dependent enzyme. A putative ‘auto-inhibitory element’, a ~50 amino acid residue segment present in the FMN-binding domain of eNOS impedes CaM binding to the enzyme, and thus high Ca2+ level is required for the enzyme activation.5, 6) While Ca2+/ CaM is a major regulator of eNOS activity, it has become evident that eNOS activity is regulated by other mechanisms as well. First, eNOS activity is negatively or positively regulated by protein-protein interactions. When eNOS is bound to caveolin-1 (Cav-1) in caveolae, its activity is repressed and this tonic inhibition can be released by displacing Cav-1 with Ca2+/CaM.7,8) Heat shock protein 90,9) a
GTP-binding protein dynamin-2,10) a voltage-dependent anion channel porin11) and NOS-interacting protein (NOSIP) have also been identified to regulate eNOS activity by protein- protein interactions.12) Post-translational modification of eNOS has also been recognized as a critical mechanism controlling eNOS activity. eNOS is subjected to myristoylation and palmitoylation which target the enzyme to the plasma membrane or Golgi.13,14) The sub-cellular locations of eNOS has been found to be critical for the enzyme activation through signal transduction.15,16)
The importance of eNOS phosphorylation has only been recognized recently.17,18) At least five specific phosphorylation sites on eNOS have been recognized; Ser114, Thr495, Ser615, Ser633 and Ser1177,19,20) numbering based on mouse eNOS sequence. Although the evidence supporting the importance of these phosphorylation sites on eNOS has been growing, there is significant controversy regarding the protein kinases and phosphatases that regulate phosphorylation of each site.21) For example, recent studies have proposed that non-receptor protein kinase Src regulates eNOS-Ser1177 phosphorylation by activating phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway in response to fluid shear stress.22,23) However, earlier studies have questioned whether Akt was responsible for eNOS activation by shear stress.24) Instead, PI3K/protein kinase A (PKA) pathway was proposed to be a more plausible mechanism.25) Therefore questions remain if these two pathways regulate eNOS independently.
Cross-talk between PKA and Src signaling has also been noticed. COOH-terminal Src kinase, which negatively
*Corresponding author
Phone: 82-53-420-4946; Fax: 82-53-427-4911 E-mail: [email protected]
Abbreviations: NO, nitric oxide; eNOS, endothelial nitric oxide syn- thase; CaM, calmodulin; Cav-1, caveolin-1; PI3K, phosphoinositide 3- kinase; Akt, protein kinase B; PKA, protein kinase A, MAEC, mouse aorta endothelial cell; DMEM, Dulbecco’s modified Eagle’s medium;
VEGF, vascular endothelial growth factor; L-NAME, nitro-L-arginine methyl ester
regulates Src activity by phosphorylating at Tyr527, has in turn shown to be activated by PKA-dependent phosphorylation at Ser364.26) PKA directly phosphorylates Src at Ser17 although the resulting effect on Src activity is controversial.27,28) Therefore, in the present study, the association of Src and PKA in eNOS regulation was examined by expressing Src and PKA mutants in cultured mouse aorta endothelial cells (MAECs).
Materials and Methods
Isolation and Culture of MAECs.
MAECs were isolated from pathogen-free male C57BL/6 mice as described previously.29) The cells were plated on 0.5% gelatin-coated 100-mm plates (Falcon) and maintained at 37oC and 5% CO2in a growth medium [Dulbecco’s modifiedEagle’s medium (DMEM) containing 20% fetal bovine serum, 100 µg/ml
endothelial cell growth supplement (Sigma), and 1% penicillin/
streptomycin]. Cells used in this study were between passages 4 and 10.
Expression Vectors.
PKA-Cqr and PKA-Rab constructs were kind gifts from Dr. G. Stanley McKnight, University of Washington. PKA-Cqr is a constitutively active mutant of mouse PKA catalytic subunit Cα which contains double mutations (His87Gln and Trp196Arg),30) and PKA-Rab is a dominant negative mutant of PKA regulatory subunit RIαwhich has mutations of Gly200Glu, Gly324Asp and Arg332His.31) The entire coding sequences of PKA-Cqr and Rab were sub- cloned into a bi-cistronic vector, pAdTrack (a kind gift from Dr. Bert Vogelstein, Johns Hopkins University) which also expresses green fluorescent protein. The pSGT vector expressing constitutively active chicken c-Src-Y527F has been described in detail elsewhere.32)
Antibodies.
The following primary antibodies were used for Western blotting: polyclonal antibodies for phosphor-Akt (Ser473), phosphor-eNOS (Ser1177), Phosphor-Src (Tyr416 and Tyr527) from Cell Signaling Technology, polyclonal antibodies for total Akt, Src, and actin from Santa Cruz Biotechnology, and monoclonal antibodies for total eNOS, PKA-Cα, PKA-RIα, and phosphor-Cav-1 (Tyr14) from BD Transduction Laboratories.Shear Stress Treatment.
A confluent MAEC monolayer grown on a 100 mm dish was exposed to non-pulsatile, laminar shear stress in a low serum-containing medium A (phenol red-free DMEM containing 0.5% fetal bovine serum and 25 mM Hepes, pH 7.4) by rotating a Teflon cone (0.5o cone angle) as described previously.24)Transfection.
MAECs were transfected with plasmid vectors using lipofectamin (Invitrogen). Cells grown to ~90%confluency in 6-well plates were washed with and kept in 1 ml
Opti-MEM (Gibco). One µg DNA and 2µg lipofectamin were mixed with 100µl Opti-MEM and the mixture was kept at room temperature for 30 min. Cells were treated with the mixture for 5 h at 37oC. The transfected cells were further incubated for 1 day. When MAECs were transfected with
pAdTrack vector, the transfected cells were conveniently monitored by fluorescence microscopy for green fluorescent protein expression, revealing typical transfection efficiency
~50% (data not shown).
NO Production.
NO released from the cells and accumulated as nitrite or nitrate (referred to as NOx) in the cell culture media was determined using an amperometric NO sensor (AmiNO700, Harvard Apparatus) as described previously33). Briefly, cells were incubated in a low serum-containing medium A with or without treatments for specified periods.The conditioned media were collected and treated with nitrate reductase to reduce nitrate into nitrite. The total nitrite was then quantified by converting it to NO in acidic potassium iodide solution where NO was continuously monitored by an NO-sensor.
Western blotting.
Cells were washed in ice-cold phosphate buffered saline and treated in lysis buffer (10 mM Tris-HCl, pH 7.6, 1 mM sodium vanadate and 1% SDS). The lysate was further homogenized by repeated aspiration through a 25- gauge needle. Protein content was measured by a Bio-Rad DC assay. Aliquots of cell lysates (20µg protein per well) were resolved on a 10% SDS-PAGE gel and proteins were transferred onto a polyvinylidene difluoride membrane (Millipore).The membrane was incubated with a primary antibody overnight at 4oC, then with a secondary antibody conjugated with alkaline phosphatase for 1 h at room temperature. The signal was detected by a chemiluminescence method.
Statistical Analysis.
Statistical analysis was performed by Student’s t-test. The values were considered to be statistically significant at p< 0.05 based on at least three independent experiments.Results
eNOS phosphorylation in MAECs
. MAECs were examined for eNOS phosphorylation at Ser1177 in response to vascular endothelial growth factor (VEGF), cAMP, and fluid shear stress. As shown in Fig. 1, all the stimuli tested increased eNOS phosphorylation in a time-dependent manner whereas total eNOS protein level remained unchanged. The results were comparable to those observed previously in bovine aorta endothelial cells.25)Active Src stimulates eNOS.
The regulation of Src activity occurs at Tyr416 and Tyr527 (based on chicken c-Src);Phosphorylation of Tyr416 in the activation loop of the kinase domain activates the enzyme while the Tyr527 phosphorylation at C-terminus leads to its inactivation.34) The dephosphorylation of Tyr527 leads to the stimulation of Src catalytic activity.
Thus substitution of Tyr527 by another amino acid residue constitutively activates c-Src as in the case of c-Src-Y527F mutant.34)
To examine the role of Src in eNOS activation, MAECs were transfected with a vector expressing a constitutively active Src mutant (c-Src-Y527F), and control cells were transfected with an empty vector. The transfected cells were
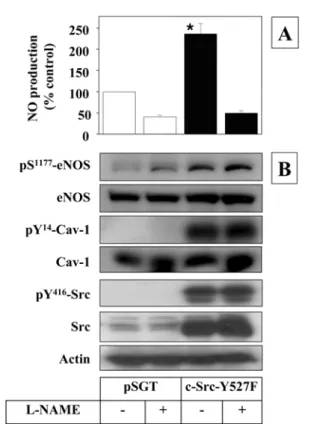
incubated in the presence and absence of nitric oxide synthase inhibitor, nitro-L-arginine methyl ester (L-NAME) for 20 h, and NO production was investigated. As shown in Fig. 2A, protein expression of c-Src-Y527F stimulated NO production which was inhibited by L-NAME.
The cell lysates were subjected to Western blotting to examine expression and phosphorylation of eNOS (Fig. 2B).
A robust expression of c-Src-Y527F was evident as compared with actin. As expected, the expressed c-Src-Y527F underwent auto-phosphorylation at Tyr416 leading to the enzyme activation. The activity of Src was further verified by the phosphorylation of Cav-1 at Tyr14, a known endogenous substrate of Src.35) The expression of active Src enhanced eNOS phosphorylation markedly without affecting the expression of eNOS. These results indicate that Src signaling is sufficient to stimulate eNOS phosphorylation and activation.
PKA-dependent eNOS regulation does not involve Src signaling.
It has been shown that PKA activation leads to the phosphorylation and activation of eNOS.33) Therefore, involvement of Src activity in PKA-dependent eNOS regulation was examined utilizing a constitutively active PKA catalytic subunit mutant (PKA-Cqr) and a dominant negative PKA regulatory subunit mutant (PKA-Rab).PKA holoenzyme is composed of two catalytic (C) subunits, and two regulatory (R) subunits.36) The enzyme activity is repressed when C subunits are bound to the inhibitory R subunits. When intracellular cAMP level increases, cAMP binds to R subunits changing their conformation. This allows the C subunits to be released from the R subunits and become catalytically active. PKA-Cqr has mutations at the binding sites for R subunits and thus is resistant to inhibition by R subunits.30) It is thus constitutively active even at the basal level of intracellular cAMP. On the other hand, cAMP cannot bind to PKA-Rab because this subunit has mutations at cAMP binding sites.31) The subunit tends to bind to C subunits even if the intracellular cAMP level is elevated. PKA-Rab mutant thus inhibits PKA activity in a dominant negative manner.
When MAECs were transfected with these constructs, the
expression of PKA-Cqr and Rab was clearly detected by antibodies for C and R subunits, respectively, as shown in Fig.
3. The actin blot served as a control. Expression of PKA-Cqr stimulated NO production and eNOS phosphorylation at Ser1177 without affecting the total eNOS protein level.
However, neither PKA-Cqr nor Rab expression affected phosphorylation status of Src at Tyr416 and Tyr527. Src- Tyr527 was highly phosphorylated while Tyr416 was minimally phosphorylated, indicating Src activity was repressed. Thus it is implicated that PKA may regulate eNOS without affecting Src signaling.
Src regulates eNOS by an Akt-dependent but PKA- independent mechanism.
Next, it was questioned whether Src signaling regulates eNOS involving a PKA-dependent mechanism. To test this, c-Src-Y527F construct or an empty pSGT vector was co-transfected with either PKA-Rab or an empty pAdTrack vector in MAECs. As shown in Fig. 4, NO production and eNOS phosphorylation stimulated by Src were Fig. 1. Phosphorylation of eNOS at Ser1177 in response toVEGF, cAMP and shear stress in MAECs. Confluent mono- layers of MAECs were exposed to VEGF (50 ng/ml), 8-Br- cAMP (1 mM) or laminar shear stress (15 dyn/cm2) for indicated time (min). Cell lysates were analyzed by Western blot with antibodies specific for phosphor-eNOS (Ser1177). The mem- branes were re-probed with antibodies detecting total eNOS to monitor equal loading of samples. Blots shown are representa- tives of at least three independent studies.
Fig. 2. Src stimulates NO production and eNOS phosphorylation in MAEC. Sub-confluent MAECs were transfected with a vec- tor expressing c-Src-Y527F or an empty pSGT vector (1µg/
well). After 24 h-incubation, the medium was changed to fresh low-serum medium containing L-NAME (1 mM) or vehicle.
The cells were incubated further for 20 h and the conditioned media were collected to measure NO production as described in Materials and Methods (panel A). NO production was normalized for total protein. Data are expressed as percent of control NO production from the cells transfected with an empty pSGT vector and treated with vehicle only (6.5 nmol/mg protein/20 h).
Data represent mean ± SEM (n = 3). *, p< 0.05 vs. vector control. The cell lysates were analyzed by Western blot using antibodies specific to the proteins indicated (panel B). Blots shown are representatives of at least three independent studies.
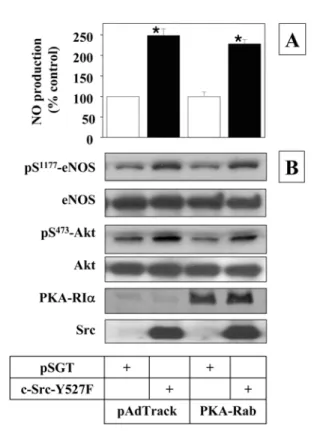
not inhibited by co-expressed PKA-Rab. This result implicates that Src regulates eNOS phosphorylation and activation by a PKA-independent mechanism. As expected, active Src stimulated Akt phosphorylation that was not affected by PKA-Rab expression, supporting a role of Akt in Src- stimulated eNOS activation (Fig. 4).
Discussion
NO production from endothelial cells is stimulated by fluid shear stress, cyclic stretching and a variety of humoral factors ranging from growth factors and peptide hormones. The mechanisms by which NO production is regulated are quite variable and eNOS activity is regulated by Ca2+-dependent or -independent mechanisms or both in response to a certain stimulus.37)
Serine/threonine phosphorylation of eNOS has been noticed as one of the major ‘Ca2+-independent mechanism’ for eNOS
activation for the past several years.21) eNOS has multiple phosphorylation sites, and each site appears to play a unique role in the regulation of eNOS. eNOS-Ser1177 has been most extensively studied and its phosphorylation is known to lead to enzyme activation. The site was initially found as an Akt phosphorylation site,17,38) but later studies showed that many other protein kinases such as protein kinase A and G,39) AMP- activated kinase40) and CaM kinase II41) also regulated the phosphorylation of this site. Therefore, it is currently viewed that eNOS-Ser1177 phosphorylation is regulated by multiple mechanisms involving different protein kinase(s) at specific cellular circumstances.
Involvement of protein tyrosine kinase in eNOS regulation by Ca2+-independent mechanism has been noticed in earlier studies; treatment of endothelial cells with tyrosine kinase inhibitors has been shown to inhibit NO production in response to shear stress.42,43) However, in other studies, tyrosine phosphatase inhibitor was shown to stimulate tyrosine phosphorylation of eNOS which was associated with a decrease in the enzyme activity due to interaction with Cav- 1.44) Fleming et al. showed that treatment of endothelial cells with a tyrosine phosphatase inhibitor, phenylarsine oxide, Fig. 3. PKA regulates eNOS without involving Src. Sub-
confluent MAECs were transfected with a vector expressing PKA-Cqr, PKA-Rab or an empty pAdTrack vector (1µg/well).
After 24 h-incubation, the medium was changed to fresh low- serum medium. The cells were incubated further for 20 h and the conditioned media were collected to measure NO production
(panel A). Data represent mean ± SEM (n = 3). *, p< 0.05 vs.
vector control. The cell lysates were analyzed by Western blot using antibodies specific to the protein indicated (panel B).
PKA-Cqr and Rab mutants were detected by the antibodies for wild type PKA-Cα and RIα, respectively. Blots shown are rep- resentatives of at least three independent studies.
Fig. 4. Src regulates eNOS activity by a PKA-independent but Akt-dependent mechanism. Sub-confluent MAECs were co-transfected with 0.5µg pSGT vectors plus 0.5µg PKA-Cqr constructs per well as indicated. After 24 h-incubation, the medium was changed to fresh low-serum medium. The cells were incubated further for 20 h and the conditioned media were collected to measure NO production (panel A). Data represent mean ± SEM (n = 3). *, p< 0.05 vs. vector control (pSGT). The cell lysates were analyzed by Western blot using antibodies spe- cific to the proteins indicated (panel B). Blots shown are repre- sentatives of at least three independent studies.
activates eNOS in a Ca2+-independent manner.45) Although the Ca2+-independent NO production was highly sensitive to tyrosine kinase inhibitors, eNOS was shown to be rapidly dephosphorylated after stimulation with phenylarsine oxide.
These observations have suggested that protein tyrosine kinases are unlikely to phosphorylate eNOS directly. Instead, certain tyrosine kinase(s) such as Src may regulate eNOS activity indirectly by controlling the signaling pathway leading to eNOS phosphorylation.
Evidence for the involvement of Src in eNOS regulation has become available recently. It has been shown that Src regulates Ser1177 phosphorylation in response to various stimuli such as hydrogen peroxide,46) estrogen,47,48) sphingosine 1-phosphate and VEGF,49) and fluid shear stress.22,23) Src is considered to regulate eNOS by activating Akt in a PI3K-dependent mechanism, although it is also known to activate Akt by direct tyrosine phosphorylation.50) The present study demonstrated that stimulation of Src signaling alone is sufficient to induce eNOS activation by an Akt-dependent mechanism, independently of PKA. Supporting this, over-expression of a constitutively active Src mutant appeared to induce eNOS-Ser1177 (Fig. 2) and Akt phosphorylation (Fig. 4).
Direct effect of Src on PKA activity was not examined in the present study because PKA has been implicated to be a putative upstream regulator, rather than a regulatory target of Src. Instead, the effect of PKA inhibition on the Src- stimulated eNOS activation was investigated. As shown in Fig. 4, Src-stimulated phosphorylation of eNOS was not affected by PKA inhibition with a dominant negative mutant.
Therefore, although it is yet unclear whether Src regulates PKA directly, PKA is not thought to play a critical role in eNOS activation by Src. Furthermore, the present study did not support the role of PKA in Src regulation, because the activation or inhibition of PKA activity had no significant effect on tyrosine phosphorylation of the activation and inactivation site in Src (Fig. 3). Based on these results, it is very likely that Src and PKA regulate eNOS independently, although additional studies using various activators or inhibitors of PKA would be necessary for a more conclusive validation.
As reported in a previous study, shear stress also stimulates eNOS-Ser1177 phosphorylation by a mechanism which is PI3K and PKA-dependent, but independent of Akt.24) Then, how is PKA activated by PI3K-dependent mechanism without involving Src or related tyrosine kinases? Class I PI3Ks expressed in endothelial cells are regulated by protein tyrosine kinase-dependent mechanism (PI3Kα, β and δ), or G protein- dependent mechanism (PI3Kγ, and β)51). PI3Kγ, which is uniquely activated by a G protein-dependent and Src- independent mechanism, has been reported to be stimulated in endothelial cells exposed to fluid shear stress.52) Then shear stress-stimulated eNOS phosphorylation might be regulated by either G protein/PI3Kγ/PKA pathway or Src/PI3Ks/Akt pathway. Further studies are imperative to delineate the signaling pathway regulating eNOS activation by shear stress.
In conclusion, this study implicates that Src/Akt pathway and PKA signaling may regulate eNOS phosphorylation independently. The existence of multiple mechanisms for eNOS phosphorylation should be beneficial to generate adequate NO production under various physiological conditions.
Acknowledgments.
This research was supported by Kyungpook National University Research Fund, 2005.References
1. Bredt, D. S. (1999) Endogenous nitric oxide synthesis: bio- logical functions and pathophysiology. Free Radic. Res. 31, 577-596.
2. Alderton, W. K., Cooper, C. E. and Knowles, R. G. (2001) Nitric oxide synthases: structure, function and inhibition.
Biochem. J. 357, 593-615.
3. Huang, P. L., Huang, Z., Mashimo, H., Bloch, K. D., Mosk- owitz, M. A., Bevan, J. A. and Fishman, M. C. (1995) Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377, 239-242.
4. Shesely, E. G., Maeda, N., Kim, H. S., Desai, K. M., Krege, J. H., Laubach, V. E., Sherman, P. A., Sessa, W. C. and Smithies, O. (1996) Elevated blood pressures in mice lack- ing endothelial nitric oxide synthase. Proc. Natl. Acad. Sci.
USA 93, 13176-13181.
5. Chen, P. F. and Wu, K. K. (2000) Characterization of the roles of the 594-645 region in human endothelial nitric- oxide synthase in regulating calmodulin binding and elec- tron transfer. J. Biol. Chem. 275, 13155-13163.
6. Nishida, C. R. and Ortiz de Montellano, P. R. (1999) Auto- inhibition of endothelial nitric-oxide synthase. Identification of an electron transfer control element. J. Biol. Chem. 274, 14692-14698.
7. Ju, H., Zou, R., Venema, V. J. and Venema, R. C. (1997) Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J. Biol. Chem. 272, 18522-18525.
8. Michel, J. B., Feron, O., Sacks, D. and Michel, T. (1997) Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+- calmodulin and caveolin. J. Biol. Chem. 272, 15583- 15586.
9. Garcia-Cardena, G., Fan, R., Shah, V., Sorrentino, R., Cir- ino, G., Papapetropoulos, A. and Sessa, W. C. (1998) Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 392, 821-824.
10. Cao, S., Yao, J., McCabe, T. J., Yao, Q., Katusic, Z. S., Sessa, W. C. and Shah, V. (2001) Direct interaction between endothelial nitric-oxide synthase and dynamin-2.
Implications for nitric-oxide synthase function. J. Biol.
Chem. 276, 14249-14256.
11. Sun, J. and Liao, J. K. (2002) Functional interaction of endothelial nitric oxide synthase with a voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 99, 13108-13113.
12. Dedio, J., Konig, P., Wohlfart, P., Schroeder, C., Kummer, W. and Muller-Esterl, W. (2001) NOSIP, a novel modulator
of endothelial nitric oxide synthase activity. Faseb. J. 15, 79-89.
13. Liu, J. and Sessa, W. C. (1994) Identification of covalently bound amino-terminal myristic acid in endothelial nitric oxide synthase. J. Biol. Chem. 269, 11691-11694.
14. Sessa, W. C., Barber, C. M. and Lynch, K. R. (1993) Muta- tion of N-myristoylation site converts endothelial cell nitric oxide synthase from a membrane to a cytosolic protein.
Circ. Res. 72, 921-924.
15. Gonzalez, E., Kou, R., Lin, A. J., Golan, D. E. and Michel, T. (2002) Subcellular targeting and agonist-induced site-spe- cific phosphorylation of endothelial nitric-oxide synthase. J.
Biol. Chem. 277, 39554-39560.
16. Fulton, D., Babbitt, R., Zoellner, S., Fontana, J., Acevedo, L., McCabe, T. J., Iwakiri, Y. and Sessa, W. C. (2004) Tar- geting of endothelial nitric-oxide synthase to the cytoplas- mic face of the Golgi complex or plasma membrane regulates Akt- versus calcium-dependent mechanisms for nitric oxide release. J. Biol. Chem. 279, 30349-30357.
17. Fulton, D., Gratton, J. P., McCabe, T. J., Fontana, J., Fujio, Y., Walsh, K., Franke, T. F., Papapetropoulos, A. and Sessa, W. C. (1999) Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399, 597-601.
18. Dimmeler, S., Fleming, I., Fisslthaler, B., Hermann, C., Busse, R. and Zeiher, A. M. (1999) Activation of nitric oxide synthase in endothelial cells by Akt- dependent phos- phorylation. Nature 399, 601-605.
19. Michell, B. J., Harris, M. B., Chen, Z. P., Ju, H., Venema, V. J., Blackstone, M. A., Huang, W., Venema, R. C. and Kemp, B. E. (2002) Identification of regulatory sites of phosphorylation of the bovine endothelial nitric-oxide syn- thase at serine 617 and serine 635. J. Biol. Chem. 277, 42344-42351.
20. Michell, B. J., Chen, Z., Tiganis, T., Stapleton, D., Katsis, F., Power, D. A., Sim, A. T. and Kemp, B. E. (2001) Coor- dinated control of endothelial nitric-oxide synthase phospho- rylation by protein kinase C and the cAMP-dependent protein kinase. J. Biol. Chem. 276, 17625-17628.
21. Boo, Y. C. and Jo, H. (2003) Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases.
Am. J. Physiol. Cell Physiol. 285, C499-508.
22. Jin, Z. G., Ueba, H., Tanimoto, T., Lungu, A. O., Frame, M.
D. and Berk, B. C. (2003) Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide syn- thase. Circ. Res. 93, 354-363.
23. Jin, Z.-G., Wong, C., Wu, J. and Berk, B. C. (2005) Flow shear stress stimulates Gab1 tyrosine phosphorylation to mediate protein kinase B and endothelial nitric-oxide syn- thase activation in endothelial cells. J. Biol. Chem. 280, 12305-12309.
24. Boo, Y. C., Sorescu, G., Boyd, N., Shiojima, I., Walsh, K., Du, J. and Jo, H. (2002) Shear stress stimulates phosphory- lation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms: role of protein kinase A. J.
Biol. Chem. 277, 3388-3396.
25. Boo, Y. C., Hwang, J., Sykes, M., Michell, B. J., Kemp, B.
E., Lum, H. and Jo, H. (2002) Shear stress stimulates phos- phorylation of eNOS at Ser(635) by a protein kinase A- dependent mechanism. Am. J. Physiol. Heart Circ. Physiol.
283, H1819-1828.
26. Vang, T., Torgersen, K. M., Sundvold, V., Saxena, M., Levy, F. O., Skalhegg, B. S., Hansson, V., Mustelin, T. and Tasken, K. (2001) Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J. Exp. Med. 193, 497-507.
27. Schmitt, J. M. and Stork, P. J. (2002) PKA phosphorylation of Src mediates cAMP's inhibition of cell growth via Rap1.
Mol. Cell 9, 85-94.
28. Abrahamsen, H., Vang, T. and Tasken, K. (2003) Protein kinase A intersects SRC signaling in membrane micro- domains. J. Biol. Chem. 278, 17170-17177.
29. Hwang, J., Saha, A., Boo, Y. C., Sorescu, G. P., McNally, J.
S., Holland, S. M., Dikalov, S., Giddens, D. P., Griendling, K. K., Harrison, D. G. and Jo, H. (2003) Oscillatory shear stress stimulates endothelial production of O2− from p47phox- dependent NAD(P)H oxidases, leading to monocyte adhe- sion. J. Biol. Chem. 278, 47291-47298.
30. Orellana, S. A. and McKnight, G. S. (1992) Mutations in the catalytic subunit of cAMP-dependent protein kinase result in unregulated biological activity. Proc. Natl. Acad.
Sci. USA 89, 4726-4730.
31. Clegg, C. H., Correll, L. A., Cadd, G. G. and McKnight, G.
S. (1987) Inhibition of intracellular cAMP-dependent pro- tein kinase using mutant genes of the regulatory type I sub- unit. J. Biol. Chem. 262, 13111-13119.
32. Erpel, T., Superti-Furga, G. and Courtneidge, S. (1995) Mutational analysis of the Src SH3 domain: the same resi- dues of the ligand binding surface are important for intra- and intermolecular interactions. EMBO J. 14, 963-975.
33. Boo, Y. C., Sorescu, G. P., Bauer, P. M., Fulton, D., Kemp, B. E., Harrison, D. G., Sessa, W. C. and Jo, H. (2003) Endothelial NO synthase phosphorylated at SER635 pro- duces NO without requiring intracellular calcium increase.
Free Radic. Biol. Med. 35, 729-741.
34. Tatosyan, A. G. and Mizenina, O. A. (2000) Kinases of the Src family: structure and functions. Biochemistry (Mosc) 65, 49-58.
35. Li, S. W., Seitz, R. and Lisanti, M. P. (1996) Phosphoryla- tion of caveolin by Src tyrosine kinases - The α - isoform of caveolin is selectively phosphorylated by v-Src in vivo. J. Biol. Chem. 271, 3863-3868.
36. Brandon, E. P., Idzerda, R. L. and McKnight, G. S. (1997) PKA isoforms, neural pathways, and behaviour: making the connection. Curr. Opin. Neurobiol. 7, 397-403.
37. Fleming, I., Bauersachs, J. and Busse, R. (1997) Calcium- dependent and calcium-independent activation of the endot- helial NO synthase. J. Vasc. Res. 34, 165-174.
38. Dimmeler, S., Dernbach, E. and Zeiher, A. M. (2000) Phos- phorylation of the endothelial nitric oxide synthase at ser-
1177 is required for VEGF-induced endothelial cell migra- tion. FEBS Lett. 477, 258-262.
39. Butt, E., Bernhardt, M., Smolenski, A., Kotsonis, P., Frohlich, L. G., Sickmann, A., Meyer, H. E., Lohmann, S.
M. and Schmidt, H. H. (2000) Endothelial nitric-oxide syn- thase (type III) is activated and becomes calcium indepen- dent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J. Biol. Chem. 275, 5179-5187.
40. Chen, Z. P., Mitchelhill, K. I., Michell, B. J., Stapleton, D., Rodriguez-Crespo, I., Witters, L. A., Power, D. A., Ortiz de Montellano, P. R. and Kemp, B. E. (1999) AMP-activated protein kinase phosphorylation of endothelial NO synthase.
FEBS Lett. 443, 285-289.
41. Fleming, I., Fisslthaler, B., Dimmeler, S., Kemp, B. E. and Busse, R. (2001) Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide syn- thase activity. Circ. Res. 88, E68-75.
42. Ayajiki, K., Kindermann, M., Hecker, M., Fleming, I. and Busse, R. (1996) Intracellular pH and tyrosine phosphoryla- tion but not calcium determine shear stress-induced nitric oxide production in native endothelial cells. Circ. Res. 78, 750-758.
43. Corson, M. A., James, N. L., Latta, S. E., Nerem, R. M., Berk, B. C. and Harrison, D. G. (1996) Phosphorylation of endothelial nitric oxide synthase in response to fluid shear stress. Circ. Res. 79, 984-991.
44. Garcia-Cardena, G., Fan, R., Stern, D. F., Liu, J. and Sessa, W. C. (1996) Endothelial nitric oxide synthase is regulated by tyrosine phosphorylation and interacts with caveolin-1. J.
Biol. Chem. 271, 27237-27240.
45. Fleming, I., Bauersachs, J., Fisslthaler, B. and Busse, R.
(1998) Ca2+-independent activation of the endothelial nitric oxide synthase in response to tyrosine phosphatase inhibi- tors and fluid shear stress. Circ. Res. 82, 686-695.
46. Thomas, S. R., Chen, K. and Keaney, J. F., Jr. (2002) Hydrogen peroxide activates endothelial nitric-oxide syn- thase through coordinated phosphorylation and dephospho- rylation via a phosphoinositide 3-kinase-dependent signaling pathway. J. Biol. Chem. 277, 6017-6024.
47. Haynes, M. P., Sinha, D., Russell, K. S., Collinge, M., Ful- ton, D., Morales-Ruiz, M., Sessa, W. C. and Bender, J. R.
(2000) Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ. Res. 87, 677-682.
48. Haynes, M. P., Li, L., Sinha, D., Russell, K. S., Hisamoto, K., Baron, R., Collinge, M., Sessa, W. C. and Bender, J. R.
(2003) Src kinase mediates phosphatidylinositol 3-kinase/
Akt-dependent rapid endothelial nitric-oxide synthase activa- tion by estrogen. J. Biol. Chem. 278, 2118-2123.
49. Tanimoto, T., Jin, Z. G. and Berk, B. C. (2002) Transactiva- tion of vascular endothelial growth factor (VEGF) receptor Flk-1/KDR is involved in sphingosine 1-phosphate-stimu- lated phosphorylation of Akt and endothelial nitric-oxide synthase (eNOS). J. Biol. Chem. 277, 42997-43001.
50. Chen, R., Kim, O., Yang, J., Sato, K., Eisenmann, K. M., McCarthy, J., Chen, H. and Qiu, Y. (2001) Regulation of Akt/PKB activation by tyrosine phosphorylation. J. Biol.
Chem. 276, 31858-31862.
51. Oudit, G. Y., Sun, H., Kerfant, B. G., Crackower, M. A., Penninger, J. M. and Backx, P. H. (2004) The role of phos- phoinositide-3 kinase and PTEN in cardiovascular physiol- ogy and disease. J. Mol. Cell. Cardiol. 37, 449-471.
52. Go, Y. M., Park, H., Maland, M. C., Darley-Usmar, V. M., Stoyanov, B., Wetzker, R. and Jo, H. (1998) Phosphatidyli- nositol 3-kinase gamma mediates shear stress-dependent activation of JNK in endothelial cells. Am. J. Physiol. 275, H1898-H1904.