First Principles Computational Study of Surface Reactions Toward Design Concepts of High Functional Electrocatalysts for Oxygen Reduction Reaction in a Fuel Cell System
9
0
0
전체 글
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
수치
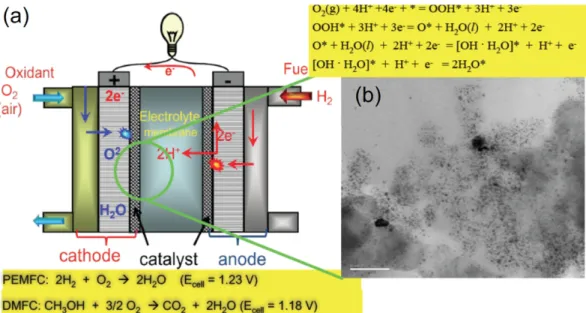
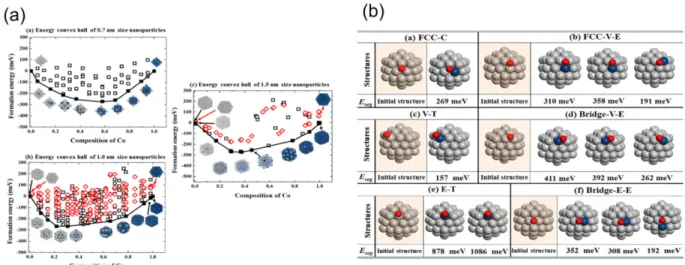
![Fig. 3. The Pourbaix diagram predicted by DFT calculations for Pt nanoparticle of 1 nm size in (a) and in (b) the in- in-situ STM measurement of electrochemical dissolution potentials of Pt with varying sizes[19].](https://thumb-ap.123doks.com/thumbv2/123dokinfo/5301511.379262/4.892.478.812.327.497/pourbaix-predicted-calculations-nanoparticle-measurement-electrochemical-dissolution-potentials.webp)

관련 문서
