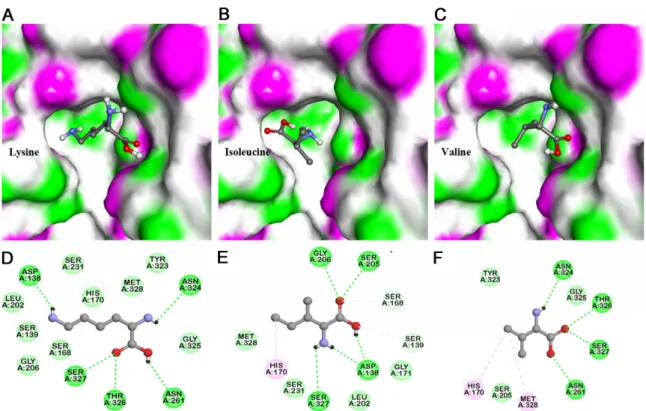
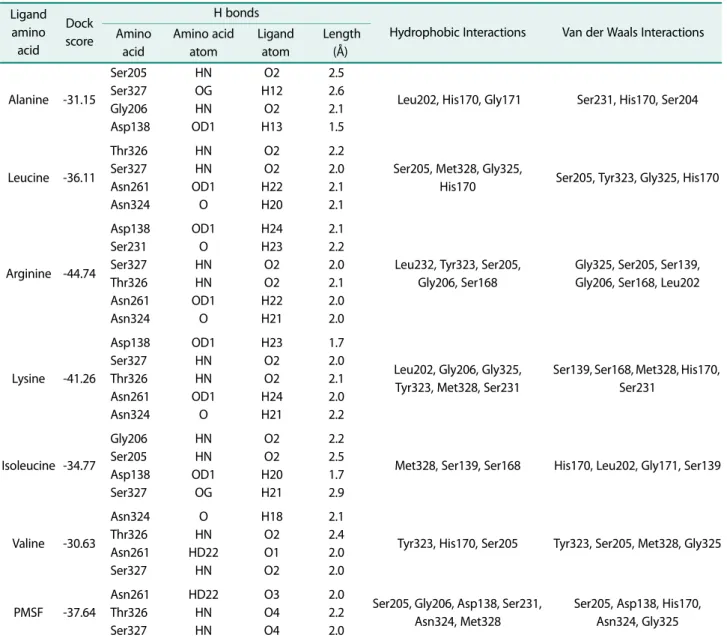
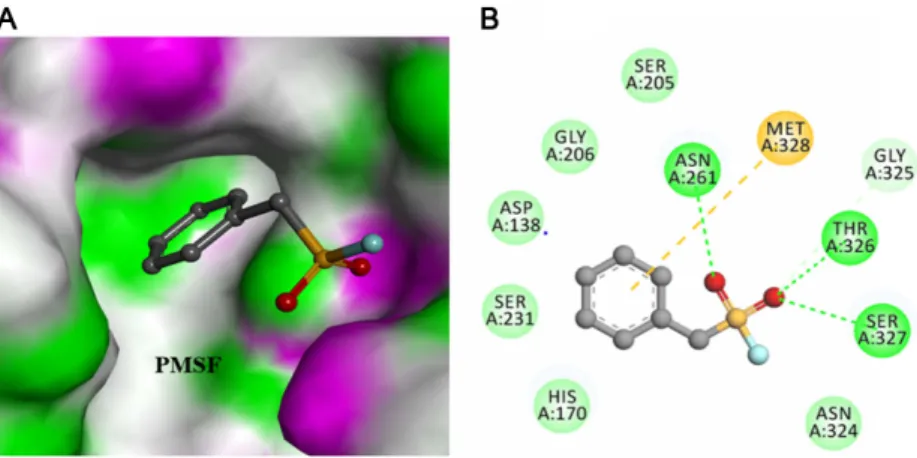
Exploring the Catalytic Significant Residues of Serine Protease Using Substrate-Enriched Residues and a Peptidase Inhibitor
Zahoor Khan
1,2, Maryam Shafique
2*, Amir Zeb
3,4, Nusrat Jabeen
1, Sehar Afshan Naz
2, and Arif Zubair
51
Department of Microbiology, University of Karachi 75270, Pakistan
2
Department of Microbiology, Federal Urdu University of Arts, Science and Technology, Karachi 75300, Pakistan
3
Division of Applied Life Science, Gyeongsang National University, Jinju 52828, Republic of Korea
4
College of Pharmacy and Graduate School of Pharmaceutical Sciences, Ewha Womans University, Seoul 03760, Republic of Korea
5