저작자표시-비영리-동일조건변경허락 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. l 이차적 저작물을 작성할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 동일조건변경허락. 귀하가 이 저작물을 개작, 변형 또는 가공했을 경우 에는, 이 저작물과 동일한 이용허락조건하에서만 배포할 수 있습니다.
이학 석사학위 논문
간세포암의 엑솜 염기서열 분석
아 주 대 학 교 대 학 원
의생명과학과/분자의학전공
간세포암의 엑솜 염기서열 분석
지도교수 우 현 구
이 논문을 이학 석사학위 논문으로 제출함.
2013년 2월
아 주 대 학 교 대 학 원
의생명과학과/분자의학전공
조 현 우
국문요약
-간세포암의 엑솜 염기서열 분석
간암 (hepatocellular carcinoma, 간세포암)은 높은 발병자 수에도 불구하고 종양 이질성이 높고 진행 양상이 복잡하여 통일된 연구를 진행하기 어렵다. 다양 한 양상의 간암에 대한 편향 없는 분석을 위해서는 모든 유전자를 함께 관찰하 는 genomewide한 연구가 필요하다. 간암의 기존 서열 변이 연구에서 잘 알려진 암 유발 유전자의 변이가 발견되었다. 이질성의 양상을 조사하고 아형에 따라 비 교 및 분석하기 위해 12 명의 간암 환자에게서 절제한 암 및 정상 간 조직을 사 용했으며, 엑솜 염기서열 분석을 통한 변이 양상과 microarray를 통한 유전자 발 현 양상을 확인하는 통합 분석을 실시했다. 유전자 발현 패턴 분석에서 두 개의 아형을 얻었으며, 이는 임상적으로 진단된 단계에 따라 구분되었다. 침윤성이 낮 은 아형 (Class 1)에서는 물질 대사와 관련된 유전자군의, 높은 아형 (Class 2)에 서는 후성유전학적 조절과 관련된 유전자군의 발현 수준이 높게 나타났다. 유전 자 변이 분석에서는 총 144개의 유전자에서 엑손 영역에 위치하며 알려진 단일 염기 다형성 목록에 없고 단백질 서열에 변화를 일으키는 암 특이적 변이가 발 견되었다. 변이가 중복 발견된 유전자로는 immunoglobulin family에 속하는HMCN1과 sodium ion channel을 구성하는 UNC80이 있었고, Sanger 염기서열
분석을 통해 검증되었다. Gene ontology를 이용한 서열 변이체의 기능 분석을 통해, 모든 환자에게서 세포 흡착 관련 유전자의 변이가 관찰되었고, 아형에 따 라 Class 1에서 세포 분화, Class 2에서 후성유전 조절 관련 유전자군에서 변이 양상에 차이를 보였다. 엑솜 염기서열 분석의 암 특이적 변이와 후성유전 결합위 치 간의 상관성을 분석한 결과 히스톤 H3k09 trimethylation 영역에서 G-protein coupled receptor 신호전달과 세포 흡착 관련 유전자의 변이가 나타났고, 히스톤
대사 등 후성유전학적 조절에 관여하는 유전자군이 공통으로 발견되었다. 다양한 진행 단계에서 얻은 간암의 서열 변이 분석에서 변이된 유전자의 기능 분석을 통해 Class 간의 공통점과 차이점을 발견할 수 있었고, 간암의 악성화에 기여하 는 유전자군의 기능을 찾을 수 있었다.
차 례
국문요약 i 차례 iii 그림 차례 v 표 차례 vi 약어 vii I. 서 론 1 A. 간암 개괄 1 B. 간암 서열변이 배경 1 C. NGS (next-generation sequencing) 개괄 2 D. 간암에 대한 NGS 연구 배경 3 E. 간암에서 chromatin remodeling complex의 중요성 3F. 목적 4 II. 연구대상 및 방법 5 A. 시료 준비 5 B. 유전자 발현 프로파일링 5 C. 엑솜 추출과 차세대 염기서열 분석 5 D. 간암에서의 염기서열 변이 프로파일링 6 E. Gene ontology 분석 7 F. PCR (polymerase chain reaction)을 이용한 검증 7 G. 후성유전 결합위치 데이터 8
H. 통계 분석 8
III. 결 과 9
B. 유전 발현 프로파일링을 이용한 간암 아형 발굴 9 C. 데이터 전처리 12 D. 변이 프로파일 15 E. 변이 프로파일의 통계 17 F. 차별 발현된 유전자의 기능적 분석 18 G. 서열변이 유전자의 기능적 특성 20 H. 재현성 있는 변이의 검증 22 I. 유전 변이체의 후성유전 결합위치와의 상관성 분석 23 IV. 고찰 26 V. 결론 30 참고문헌 31 부 록 45 ABSTRACT 51
그림 차례
Fig. 1. Gene expression profiling and grouping 11 Fig. 2. Filtering process 16 Fig. 3. Mutation statistics 17 Fig. 4. Enrichment plot of Class 1 and mutated genes overlapping the gene ontology entry 19 Fig. 5. Enrichment plot of Class 2 and mutated genes overlapping the gene ontology entry 19 Fig. 6. Heat map of gene ontology entries classified by gene expression
classes 21
Fig. 7. Heat map of gene ontology terms in the region labeled with histone
H3k09me3 24
Fig. 8. Heat map of gene ontology terms in the region labeled with histone
표 차례
Table 1. Clinical features of patients 10 Table 2. Base quality by read positions 13 Table 3. List of sample IDs and mapping statistics 14 Table 4. Built-in filtering criterion of GATK 15 Table 5. IGV and Sanger sequencing validation of recurrent mutations 22 Appendix 1. 149 cancer-specific mutations by exome sequencing 45
약 어
ANNOVAR. Annotate variation. BWA. Burrows-Wheeler aligner. ChIP-seq. Chromatin immunoprecipitation followed by sequencing. COSMIC. Catalogue of Somatic Mutations in Cancer. DAVID. Database for annotation, visualization, and integrated discovery. EGF. Epidermal growth factor. GATK. Genome analysis toolkit. GSEA. Gene set enrichment analysis. HCC. Hepatocellular carcinoma. IGV. Integrated genomics viewer. MAPK. Mitogen-activated protein kinase. NCBI. National Center for Biotechnology Information. NGS. Next-generation sequencing. PCR. Polymerase chain reaction. SNP. Single-nucleotide polymorphism. UCSC. University of California at Santa Cruz. VCF. Variant call format.
I. 서 론
A. 간암 개괄
간세포암 (hepatocellular carcinoma, 간암)은 간에서 발생하는 종양 중 가장 흔하게 발견되며 복잡한 요인에 의해 발병한다. 2008년 한 해에 70만 명이 간암 으로 사망하며, 세계적으로 볼 때 발병 횟수로 5위에 달한다 (Jemal 등, 2011). 원인으로는 복잡하고 다양한 요소가 있으며, 특히 간암 바이러스 B나 C의 감염 (Ye 등, 2010; McGivern과 Lemon, 2011; Michielsen과 Ho, 2011; Jiang 등, 2012; Sung 등, 2012), 음식 내 독소인 aflatoxin B1, 습관성 알코올 소비 등이 거론된 다 (American Cancer Society, 2012). 이들은 간 내 염증을 악화시키며 간암 발 달에 기여하는 것으로 생각된다 (Matsuzaki 등, 2007).B. 간암 서열변이 연구의 배경
간암에 대한 분자생물학적 연구를 통해 TP53, CTNNB1, AXIN1, IGF2R
(De Souza 등, 1995; Yamada 등, 1997), SMAD2와 SMAD4, RB1, p16-INK4A
(Kita 등, 1996; Chaubert 등, 1997) 등의 변이가 발견되었다 (Qin과 Tang 2002; Debuire와 Lemoine 2001). TP53은 가장 잘 알려진 암 억제 유전자이며, DNA 손상, 세포주기 조절 등 다양한 기능에 관여한다. 특히 TP53의 코돈 249번의 R249S 변이는 aflatoxin과 관련이 있으며 (Bressac 등, 1991), 중국과 아프리카 지역에서 발생한 간암의 각 각 36%, 32%에서 발견된다 (Murakami 등, 1991). 또한 전세계적으로는 진단된 간암의 11%에서 발견된다 (Puisieux와 Ozturk, 1997). Wnt 세포 신호전달 경로 의 전달 물질인 β-catenin (CTNNB1)의 변이는 간암의 20-25%에서 발견되며 (de La Coste 등, 1998; Miyoshi 등, 1998), β-catenin의 scaffold 기능을 하는
growth factor)-β 신호전달 경로의 조절자이자 암 억제 유전자인 SMAD2와
SMAD4의 변이율은 전체의 10% 미만이다 (Kawate 등, 1999; Yakicier 등,
1999). TP53의 downstream target이며 세포주기를 조절하는 유전자인 RB1의 변 이는 약 15%의 간암에서 나타난다 (Nakamura 등, 1991). Cyclin D1 (CCND1) 역시 세포주기 조절자이며, 간암의 10-20%에서 과발현이 나타난다 (Nishida 등, 1994). 하지만 이러한 연구는 이미 알려진 유전자를 중심으로 한 연구이므로 새 로운 표적 유전자를 찾아내기 위해서는 genomewide한 방법이 필요하다.
C. NGS (next-generation sequencing) 개괄
전장 유전체의 염기서열 분석을 실시하면 유전 정보의 변이를 폭넓게 관찰 할 수 있다. 염기서열 분석은 최근의 차세대 염기서열 분석 기술로 인해 분석의 속도와 비용 효율성이 증대되었다 (Wheeler 등, 2008). NGS 분석 기술은 분석하 고자 하는 DNA를 200 bp 미만의 짧은 조각으로 나누고 양을 증폭한 다음 표준 염기서열에 맞추고, 각 위치별 염기 서열을 통계적 방법으로 정하는 방식이다. NGS는 표준 염기서열과의 비교를 통해 변이를 찾아내며, 찾아낼 수 있는 변이 는 단일염기 치환 (single-nucleotide variation), 짧은 염기서열의 삽입이나 삭제 (indel), 비정상적인 splicing이나 접합이 일어난 RNA 전사체, 표준 서열과 다른 순서로 배열된 염색체 (chromosomal rearrangement) 등이 있다. 짧은 조각은 표 준 서열의 특정 위치에 유일하게 정렬되지 않을 가능성이 있으므로, paired-end 염기서열 분석을 이용해 정렬의 정확도를 높일 수 있다. 이러한 NGS 분석을 통해 새로운 체세포 변이와 유전체 재배열이 발굴되었 다. 엑솜 염기서열 분석 (Ng 등, 2010)은 단백질을 암호화하는 영역만을 추출하 여 서열을 읽는 실험으로, 더 적은 비용으로 단백질 서열에 영향을 주는 변이를 찾아낼 수 있다. 종양 유전체 연구에서는 신장암 (Xu 등, 2012), 췌장암 (Furukawa 등, 2011; Wu 등, 2011; Wang 등, 2012), 유방암 (Jiao 등, 2012), 골수암 (Hou 등, 2012), 혈액암 (Puda 등, 2012; Wang 등, 2011; Quesada 등, 2012) 등에서 엑솜 분석이 행해졌고 새로운 변이가 보고되었다. 이렇게 암에서 발견된 체세포 변이는 COSMIC (Catalogue of Somatic Mutations in Cancer)에 데이터베이스로 구축되어 있으며, 현재 약 54만 개의 암 샘플에서 136,000개의 유전자 내 서열 변이를 이용할 수 있다 (Forbes 등, 2011).
D. 간암에 대한 NGS 연구 배경
간암 유전체 NGS 연구의 첫 발표는 단일 간암 샘플에 대한 전장 유전체 해 독 (Totoki 등, 2011)이 있었고, 엑솜 염기서열 분석의 결과도 발표되었다 (Guichard 등, 2012; Li 등, 2011; Totoki 등, 2011; Zhao 등, 2011). Chromatin 구 조의 변경에 관여하는 ARID2 유전자의 기능을 상실하는 변이가 HCV와 관계된 간암 샘플에서 발견되었다 (Li 등, 2011; Zhao 등, 2011). 이후 B형 간염 바이러 스의 영향으로 인한 숙주 유전체의 광범위한 변화 (Jiang 등, 2012; Sung 등, 2012)가 발표되었고, CD90+ type 간암 세포주의 전사체 염기서열 분석이 RNA-sequencing으로 이루어졌다 (Ho 등, 2012). 유전체에 침투하는 HBV의 영 향을 분석한 논문에서 TERT 유전자와의 결합, EGF 수용체로 신호를 전달하는 ERRFI1 유전자의 변이가 다수 샘플에서 관찰되었으며 (Fujimoto 등, 2012), 또 한 한 명의 환자에서 전이된 암 조직과 주변의 정상 조직을 대상으로 엑솜 염기 서열 분석을 실시해 암의 발달 과정을 추적한 논문에서는 β-catenin이 관여하는 Wnt 신호전달 과정, MAPK 신호전달 등이 영향을 받는다 (Tao 등, 2011).
E. 간암에서 chromatin remodeling complex의 중요성
2012년에 HBV에 감염되어 발생한 간암에 대한 엑솜 염기서열 분석의 연구 결과가 발표되었다 (Huang 등, 2012). 정상 간 및 간암 조직, 그리고 간문맥으로 전이된 조직의 염기서열을 해독했으며, 총 331개의 단일염기 치환과 24개의 indel
을 발견했다. NGS 분석으로 발굴한 암 유발 유전자를 검증하기 위해 110개의 샘플에서 각 유전자의 서열을 분석한 결과, 이 중 ARID2는 HBV에 의한 간암과 HCV에 의한 간암에서 각각 변이가 확인되며, TP53의 변이는 전체 110개 샘플 중 35개, ARID1A의 변이는 14개에서 발견되었다. ARID1A의 변이는 PLC/PRF/5, Hep3B, YY-8103, HCC-LM6 등 4개의 세포주에서도 발견되었다. 특히 폐로의 전이성이 높은 HCC-LM6 세포주의 ARID1A 서열에서는 in-frame indel 1개, missense 변이 2개, 그리고 63개의 silent 변이가 발견되었다.
ARID1A는 SWI/SNF (Switch/sucrose nonfermemtable) chromatin remodeling
complex를 구성하며, 이 복합체는 전사되는 영역의 활성화를 조절한다. 이와 같 이 chromatin remodeling과 같은 후성유전학적 변화는 간암과의 상관성이 높으 며, 중요한 biomarker가 될 가능성이 높다.
F. 목적
간암의 이질성에도 불구하고 공통된 진행 양상이 나타나는 것을 감안하면 다양한 발달 단계에 해당하는 간암 조직을 이용하여 여러 단계의 방법으로 함께 분석하는 통합 분석이 필요하다. 저자는 microarray를 이용한 유전자 발현 프로 파일링과 엑솜 염기서열 해독을 통해 체세포 변이를 프로파일링하고, 간암의 진 행 단계에 따른 양상의 차이를 조사하고자 했으며, 이를 통해 암호화 영역에서 아직 알려지지 않은 체세포 변이를 발견하고, 이질성으로 나타나는 아형을 조사 하여 차이가 있는 변이 양상을 비교 분석하고자 한다.II. 연구대상 및 방법
A. 시료 준비
12명의 환자에게서 간 종양 절제술을 시행하였다. 수술은 아주대학교 병원에 서 실시되었으며, TNM staging system에 의한 종양의 발달 단계 표기에 따라 T2, T3, T4로 분류하였다. 절제된 조직에서 각각 암세포와 주변 정상 세포를 대 상으로 유전체 DNA를 추출하였다. 유전체 연구에 대한 윤리 규정을 준수하였다.B. 유전자 발현 프로파일링
Illumina TotalPrep 96 RNA 추출 키트를 제조사 제공 protocol에 맞게 사용 했다. 추출된 RNA는 Illumina HumanHT-12 v4 Expression BeadChip Kit를 사 용해 hybridization되었으며 Illumina GenomeStudio 소프트웨어를 이용해 각 유 전자의 발현양이 측정되었다. 샘플간 동일 조건의 비교를 위해 데이터에 quantile normalization을 적용했으며 Java TreeView (Saldanha, 2004) 소프트웨어를 이용 해 발현양의 차이를 heat map으로 도시하였다. 통계적 유의성을 갖는 unsupervised clustering으로 유전자 발현 프로파일에 따른 아형을 분류하기 위 해 R package pvclust (Suzuki와 Shimodaira, 2006)가 사용되었다.
C. 엑솜 추출과 차세대 염기서열 분석
엑솜 분석을 위해 Illumina TruSeq 엑솜 추출 키트를 제조사 제공 protocol 에 따라 사용하였으며, 전장 유전체의 2 %에 해당하며 20,846개의 유전자에 위 치한 62,085,295 bp를 포함하는 엑솜을 얻었다. 염기서열 해독은 Illumina HiSeq 2000 기기를 이용했으며, 왼쪽과 오른쪽의 read가 일정 간격을 두고 떨어져 있는 paired-end 방식으로 시행했다. 해독시 각 서열 조각의 길이는 110 bp였으며, 염
기별 평균 coverage는 30X가 되도록 하였다. 염기서열 해독의 결과는 FASTQ 형식으로 저장되었다. 전반적으로 조각의 3’ 말단 부위에서 phred 점수로 표현되는 서열의 신뢰성이 감소하였는데, 각 염 기의 위치별 신뢰성은 5’ 말단으로부터 75 bp까지만 phred 점수 30 이상을 유지 했으므로, 5’ 말단으로부터 75 bp인 영역만 남기고 뒷부분은 잘라내었다.
D. 간암에서의 염기서열 변이 프로파일링
BWA (Burrows-Wheeler Aligner) 0.6.1 판 (Li 와 Durbin, 2009)을 사용하 여 염기서열 해독 데이터를 UCSC Genome Browser에서 제공하는 hg19 (GRCh37) 표준 인간 염기 서열에 맞추었고, 정렬의 정확도를 계산하여 정확도 값이 0인 조각은 이후 분석에서 제외하였다. GATK (McKenna 등, 2010; DePristo 등, 2011)의 local realignment walker를 이용해 indel의 위치를 정확히 찾았고, recalibration walker를 이용해 Illumina HiSeq 방식의 NGS 기기에서 발 생하는 신뢰성 score의 차이를 보정했다. GATK 자체 필터를 사용하여 변이의 신뢰도가 낮은 것을 제거한 후, unified genotyper v3 기능을 사용하여 각 염기서 열의 variant calling을 실시하였다. 표준 염기서열과 다르면서 신뢰성이 높은 variant를 VCF 형식으로 출력했고, 염색체상 위치로 표시된 정보의 주석은 ANNOVAR (Wang 등, 2010) 프로그램을 사용했다. ANNOVAR는 내부 데이터 베이스를 이용해 변이가 위치한 유전자의 이름과 함께 알려진 단일염기 변이에 포함되는지 등을 검색한다. 먼저 segmental duplication 영역에 위치하여 mapping된 위치의 신뢰성이 낮은 variant는 사용하지 않았다. 다음으로 엑손 영 역에 해당하는 부분만 취하고 인트론이나 유전자 외 영역, noncoding RNA, 번역 되지 않는 영역 (untranslated region, UTR)은 버렸다. 정렬이 이루어진 조각을 대상으로 각각의 염기에서 표준 서열과 일치하는 수와 일치하지 않는 수를 조사 하여, 일치하지 않는 조각이 10개 이상인 경우만 취했다. Joint-SNV-Mix (Roth
등, 2012) 소프트웨어를 사용해 유전체 정보를 담은 자료에서 각 변이의 암 특이 성 확률을 계산하고, 그 중 이종성 (heterozygous) 변이, 동종성 (homozygous) 변이, 그리고 이종성 소실 (loss of heterozygosity)에 해당하는 3가지만이 종양 특이적인 변이인 것으로 판단하여 이후 분석에 사용했다. Indel의 경우 IGV 소프 트웨어 (Thorvaldsdottir 등, 2012)를 이용해 직접 연 후 정상 간 조직에서 변이 가 전혀 발견되지 않는지를 확인했다. 추가로 알려진 dbSNP에 수록되었거나 단 백질 서열에 영향을 주지 않는 silent 변이를 제거했다.
E. Gene ontology 분석
Gene ontology 분석은 유전자 발현 연구에서 양 아형간에 차별 발현된 유전 자군과 염기서열 분석에서 확인된 유전자군을 대상으로 시행했다. 차별 발현된 유전자군은 Gene Set Enrichment Analysis (GSEA) (Subramanian 등, 2005) 소 프트웨어를 사용해 Kolmogorov-Smirnov test를 시행했고, 변이가 발견된 유전 자군은 Database for Annotation, Visualization, and Integrated Discovery (DAVID) (Huang 등, 2009) 소프트웨어를 사용해 Fisher exact test를 시행했다. Gene ontology 항목 중에서 인간 종의 biological process에 속하는 항목만을 사 용했다.F. PCR (polymerase chain reaction)을 이용한 검증
NGS 분석 결과를 검증하기 위해 체세포 변이를 포함하는 유전체에 대해 PCR을 시행했다. Gel 전기 영동으로 분리된 DNA 중 실제 결과물 영역만을 잘 라내어 Sanger sequencing을 실시했다. 여러 샘플에서 변이가 중복 발견된 유전
자인 HMCN1과 UNC80을 대상으로 시행했으며, HMCN1은 5’-AAA TTG
GCA TTT TTC TAC ATA CG-3’을 sense, 5’-TTT GGT CCA GAA GTC TCA TCA-3’을 antisense primer로 사용했다. UNC80의 693번 코돈을 검증하기
위한 primer는 5’-GGA AGA GGG AGT GCA GGT AA-3’을 sense, 5’-TTG GAA AGC TCC CTC ACT TG-3’을 antisense로 사용했으며, 1147번 코돈을 검증하기 위한 primer는 5’-TCA GGT CAA ATT CAC TAG TGC TG-3’을 sense, 5’-CAG TGG TAA AAG GCA TGT ATG A-3’을 antisense로 사용하였 다. PCR 실험의 온도는 95℃에서 5분간 변성한 후, 95℃에서 30초간 denaturation, 60℃에서 1분간 annealing, 72℃에서 2분간 extension을 유지하는 주기를 30회 반복한 후 72℃에서 5분간 final extension을 시행했다.
G. 후성유전 결합위치 데이터
UCSC Genome Browser에서 간 세포주 중의 하나인 HepG2의 후성유전 marker를 대상으로 Broad Institute가 실시한 ChIP-seq의 결과를 얻었다 (Raney 등, 2011). 변형 히스톤 H2A.Z, 히스톤 H3의 acetylation과 methylation, 그리고 CpG island 영역 등 총 12개 항목을 이용했다. 각 샘플별로 10개 이상의 조각에 서 변이가 관찰된 유전자 목록을 얻은 후 label과 겹치는 유전자군만을 선별하여 gene ontology 항목을 분석하였다.
H. 통계 분석
유전자 발현 프로파일 아형과 TNM staging system 상의 초기 및 말기 간 암의 관계를 알아보기 위해 two-way χ2 검정을 시행했다.III. 결 과
A. 간암 환자군의 임상적 특성
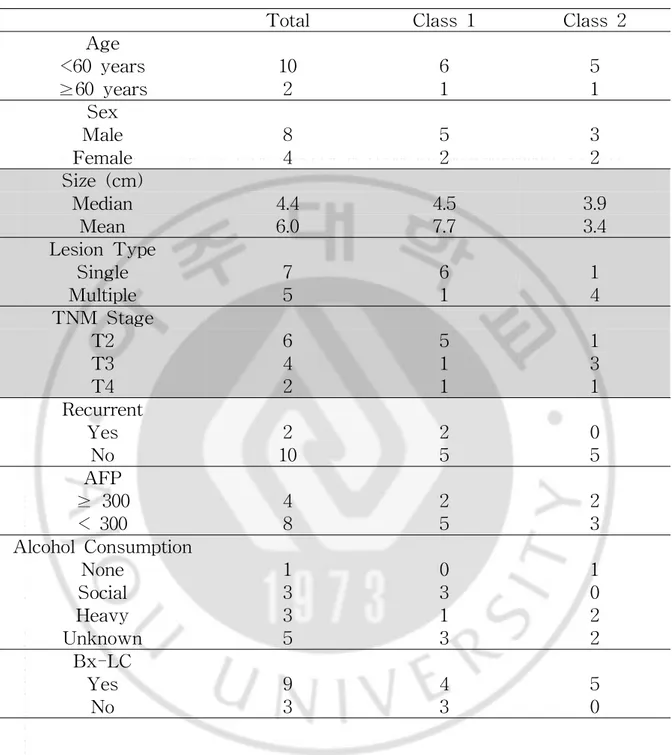
임상 데이터는 아주대학교 병원에서 기록되었다 (Table 1). 12명 중 2명 (17%)의 환자가 60세 이상이었고 4명 (33%)은 여성이었다. 절제된 암 조직 크기 의 중앙값은 4.4 cm였으며 평균값은 6.0 cm였다. 7명 (58%)의 환자에게서 단일 병변이, 5명 (42%)에게서 다중 병변이 발견되었다. 2명 (17%)의 환자는 재발한 종양을 절제하였고, 암의 지표로 쓰이는 alpha-fetoprotein (AFP)의 농도는 4명 (33%)에게서 300 ng/mL 이상으로 측정되었다.B. 유전 발현 프로파일링을 이용한 간암 아형 발굴
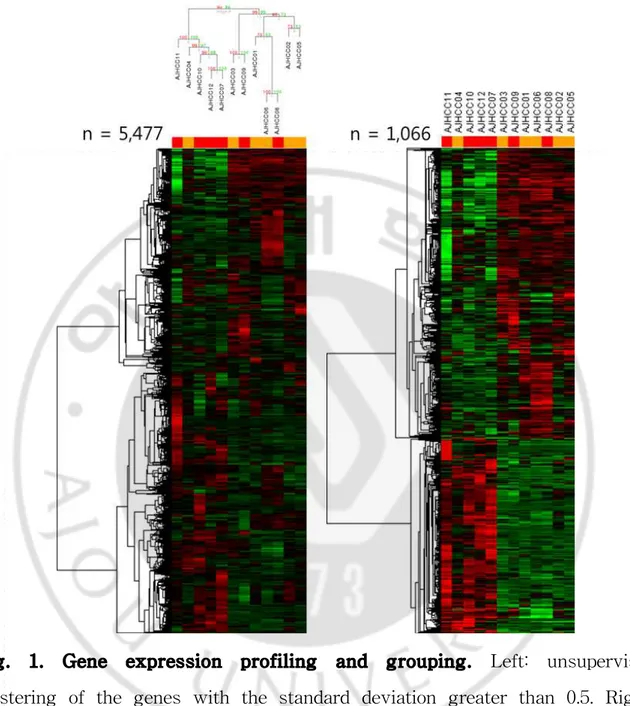
유전체 발현 프로파일을 이용해 12개 샘플의 유사성을 중심으로 2개의 아형 을 발굴하였다. Illumina Genome Studio에서 나온 발현 데이터는 변환된 다음 종양 조직의 값에서 정상 조직의 값을 뺀 수치가 사용되었다. 각 샘플별로 표준 편차 값이 0.5 이상인 유전자만을 사용했으며, R 패키지인 pvclust를 사용해 unsupervised hierarchical clustering을 실시한 결과 Class 1과 2로 유의하게 분 류되었다 (Fig. 1). Class 1은 7개, Class 2는 5개의 샘플을 포함하였고, Class 1 에서 7개 중 5개의 T2가, Class 2에서 5개 중 4개의 T3, T4 샘플이 포함되었다. χ2 검정에서는 0.07의 p 값을 얻었다. 모집단의 수가 적어 통계적 유의성은 없었 으나, Class 2에 속한 샘플의 크기가 더 작았고, TNM stage T3 및 T4에 더 많 이 속했으며, 다중 병변이 더 많이 발견되었다.Total Class 1 Class 2 Age <60 years 10 6 5 ≥60 years 2 1 1 Sex Male 8 5 3 Female 4 2 2 Size (cm) Median 4.4 4.5 3.9 Mean 6.0 7.7 3.4 Lesion Type Single 7 6 1 Multiple 5 1 4 TNM Stage T2 6 5 1 T3 4 1 3 T4 2 1 1 Recurrent Yes 2 2 0 No 10 5 5 AFP ≥ 300 4 2 2 < 300 8 5 3 Alcohol Consumption None 1 0 1 Social 3 3 0 Heavy 3 1 2 Unknown 5 3 2 Bx-LC Yes 9 4 5 No 3 3 0
Table 1. Clinical features of patients.
Two of the patients were over the age of 60, and four of them were female. Overall, samples in Class 2 showed smaller sizes, exhibited higher TNM stages, and had multiple lesions. AFP, Alpha-fetoprotein in nanograms per milliliter.
Fig. 1. Gene expression profiling and grouping. Left: unsupervised clustering of the genes with the standard deviation greater than 0.5. Right: the 1,066 differentiallly expressed genes with the expression level greater than twofold classified by the result of the unsupervised clustering. The orange color is the clinically labeled T2, and the red color T3 and T4 in the TNM staging system.
C. 데이터 전처리
엑솜 염기서열 분석은 FASTQ 형식의 데이터로 출발했다. 염기서열의 신뢰 성은 phred score로 표시되며, 0에서 40까지의 범위를 갖는다. Phred score Q는 해당 서열이 틀릴 수 있는 확률 P로부터 Q = -10log10P의 식으로 계산된다. 각
샘플의 염기서열 위치별 신뢰성을 도시한 결과 75번째 서열 이후에는 평균 신뢰 성이 떨어짐이 관찰되었으므로, 신뢰성이 높은 5’ 말단의 75 bp만이 분석에 사용 되었다 (Table 2). 준비된 데이터를 BWA를 이용해 hg19 표준 염기서열에 mapping한 결과 99 % 이상의 조각이 mapping되었으며, 그 중 평균 43 %가 exome enrichment kit의 영역에 위치했다 (Table 3).
Position QualityMean MedianQuality QuartileLower QuartileUpper Percentile10th Percentile90th 1 37.81439 39 38.31618 39 35.65395 39 2 37.66665 39 38.17927 39 35.38305 39 3 37.63082 39 38.11032 39 35.38305 39 4 37.58794 39 37.98993 39 35.36052 39 5 37.58423 39 38.08779 39 35.34186 39 6 37.61312 39 38.13538 39 35.38305 39 7 37.58061 39 37.91132 39 35.21127 39 8 37.54516 39 37.84809 39 35.04259 39 9 37.52389 39 37.74915 39 34.97042 39 10-14 37.43889 39 37.65096 39 34.75744 39 15-19 37.26523 39 37.49764 39 34.25815 39 20-24 37.00869 39 37.17339 39 33.47274 39 25-29 36.68834 38.96421 36.71052 39 32.74475 39 30-34 36.56507 38.90988 36.59517 39 32.44822 39 35-39 36.44442 38.92867 36.52212 39 32.23803 39 40-44 36.16234 38.83568 36.34859 39 31.5048 39 45-49 35.75087 38.44318 35.92788 38.97461 30.3389 39 50-54 35.49644 38.29563 35.74505 38.95769 29.0678 39 55-59 35.28155 38.27376 35.63326 38.97944 28.46195 39 60-64 34.84536 37.96686 35.21184 38.95769 27.38184 39 65-69 34.26321 37.5971 34.44496 38.94077 25.71362 39 70-74 33.51244 37.27267 33.54524 38.7457 23.18231 38.99154 75-79 33.17379 37.29528 33.05936 38.81393 20.7114 38.95769 80-84 32.64633 37.05238 32.06267 38.83922 17.18559 38.95769 85-89 31.73088 36.45021 30.86358 38.73286 10.12129 38.92021 90-94 30.65603 35.88838 29.44461 38.44881 3.341329 38.90692 95-99 29.38968 34.99402 27.2549 38.08118 2 38.86943 100-104 27.85893 33.54049 23.44393 37.38387 2 38.78421 105-109 26.06434 32.46087 16.68107 36.49842 2 38.54168 110 24.57377 30.89655 9.668787 36.1284 2 38.29273
Table 2. Base quality by read positions.
The base quality, in the range between 0 and 40, must be high to ensure confidence in sequence alignment. After the 75th base position, the quality is evidently lower than that in the the 5' end position.
Table 3. List of sample IDs and mapping statistics. Sample ID Sequenced Amount (bp) Read Length (bp) Mapped (%) On Target (bp) Average Coverage (X) ≥1X Coverage (%) ≥10X Coverage (%) AJHCC01 2,375,819,550 75 99.12 995,114,844 16.03 91.1 64.2 AJHCC02 2,396,746,950 75 99.04 1,048,782,870 16.89 91.6 67.2 AJHCC03 1,917,045,750 75 98.49 861,476,036 13.88 91.7 58.5 AJHCC04 2,642,532,900 75 99.21 1,145,827,171 18.46 91.8 70.5 AJHCC05 2,206,402,800 75 99.14 917,550,337 14.78 91.1 62.0 AJHCC06 1,936,790,100 75 99.09 857,492,245 13.81 91.7 61.1 AJHCC07 1,925,189,250 75 99.22 884,227,711 14.24 89.6 58.2 AJHCC08 1,900,917,150 75 98.93 842,934,665 13.58 91.9 61.0 AJHCC09 2,605,835,700 75 99.04 1,127,964,119 18.17 90.7 66.2 AJHCC10 2,694,500,250 75 99.02 1,170,699,176 18.86 90.7 67.1 AJHCC11 2,262,746,850 75 99.16 1,015,204,395 16.35 91.1 65.3 AJHCC12 2,624,237,250 75 99.15 1,099,207,244 17.7 91.5 66.8 AJHN01 1,855,769,400 75 99.18 782,590,154 12.61 90.5 57.6 AJHN02 2,244,950,550 75 99.1 889,886,915 14.33 90.9 62.0 AJHN03 1,970,915,700 75 99.15 847,755,822 13.65 90.1 59.6 AJHN04 2,250,287,100 75 99.26 935,558,350 15.07 90.8 63.7 AJHN05 1,914,969,600 75 99.19 798,201,711 12.86 90.1 56.9 AJHN06 2,157,990,600 75 99.14 930,842,708 14.99 89.5 59.3 AJHN07 2,544,481,800 75 98.56 1,107,361,542 17.84 92.9 68.6 AJHN08 2,185,490,550 75 99.11 933,406,308 15.03 91.1 63.9 AJHN09 2,144,887,350 75 99.1 922,953,284 14.87 90.5 62.2 AJHN10 2,191,735,650 75 99.15 951,322,941 15.32 91.3 63.7 AJHN11 2,004,286,500 75 99.16 871,387,451 14.04 89.6 58.8 AJHN12 1,770,054,600 75 99.11 777,367,668 12.52 90.7 56.6
More than 99% of the sequence data were mapped to the reference genome, and 43% mapped to the target exonic regions on average. The average coverage was 15.7X.
D. 변이 프로파일
GATK의 표준 분석 절차와 자체 필터를 (Table 4) 사용해 조각의 위치 정 보와 신뢰성 점수가 보정된 131,947개의 위치에 일어난 서열 변이 자료를 얻었 다. ANNOVAR를 사용하여 신뢰성이 낮은 서열 변이를 제거하였고 (Fig. 2), 총 144개 유전자에 속한 149개 위치에서 종양 특이적이며 단백질 서열을 바꾸는 서 열 변이 자료를 얻었다 (부록 1).Table 4. Built-in filtering criterion of GATK.
Filter criterion Filter name MQ0 ≥ 4 and MQ0 / DP > 0.1 Hard to validate
DP < 5 Low coverage QUAL < 30.0 Very low quality 30.0 < QUAL <50.0 Low quality
QD < 1.5 Low quality-by-depth
GATK provides a built-in filter that reads the statistics of a VCF file. Filters were applied as suggested by developers of GATK. Variants that are hard to validate, have low coverage, low quality, or low quality by depth were labeled as in the Table, and filtered out. MQ0, count of zero mapping quality. DP, depth. QUAL, variant quality. QD, quality by depth.
Fig. 2. Filtering process. The list of tumor-specific exonic variants were made from the results of genotyping by GATK. Variants located in regions exhibiting segmental duplication were removed. The exonic and splice site variants were chosen, and high-quality variants were picked with the criterion of more than 10 mutated reads in each chromosomal position. The software Joint-SNV-Mix pinpointed cancer-specific variants by examining the paired sequence reads of tumor and paired normal samples and calculated the probability of cancer specificity. Finally, known variants and synonymous variants were excluded to generate the final list of mutation.
E. 변이 프로파일의 통계
1,209개의 종양 특이적 변이와 그 중 319개의 알려진 SNP을 제외한 집합을 대상으로 조사했으며, Class 1과 Class 2에서 발견된 것, 공통으로 발견된 것 등 으로 나누어서 분류했다. 1,209개 집합에서는 796개의 transition과 350개의 transversion, 14개의 insertion, 49개의 deletion이 보고되었고, SNP를 제외한 319 개 집합에서는 165개의 transition, 129개의 transversion, 7개의 insertion, 18개의 deletion이 보고되었다. 1,000 Genomes Project에 의하면 인간 유전체에서 transition과 transversion의 비는 2:1로 예측되며, 단백질 암호화 영역에서는 이 보다 조금 높다 (Li 2011). 각 아형별로 계산된 transition과 transversion의 비는 알려진 SNP을 제외하는 전후로 차이를 보였으나, Class 간에 유의한 차이는 나 타나지 않았다 (Fig. 3).
Fig. 3. Mutation statistics. The transition to transversion ratio did not show significant difference between the classes.
F. 차별 발현된 유전자의 기능적 특성
Microarray 분석에서 얻은 유전자 발현 프로파일에 GSEA를 사용하여 차별 발현된 유전자군의 기능을 분석했다. 발현양 데이터와 Class 1, 2에 대한 정보를 이용하여, Class 1 (Fig. 4)에서 물질 대사 관련 ontology 항목을, Class 2 (Fig. 5)에서는 후성유전학적 변형 관련 항목을 얻었다. 발현 데이터의 기능을 볼 때, 양 아형은 침윤성의 정도에 따라 구분됨을 알 수 있다.
Fig. 4. Enrichment plot of Class 1 and mutated genes overlapping the gene ontology entry.
Class 1 showed enrichment in the various metabolic processes.
Fig. 5. Enrichment plot of Class 2 and mutated genes overlapping the gene ontology entry.
Class 2 showed enrichment in the functions related to tumor aggressiveness, such as epigenetic regulation, cell cycle, and cell adhesion.
G. 서열변이 유전자의 기능적 특성
엑솜 염기서열 분석을 통해 발견된 유전자 변이는 각각 포함되는 gene ontology 항목으로 변환 후 분석하였다. 유전자가 속한 gene ontology 항목에 관 한 데이터는 NCBI에서 얻었다. 양 Class별로 2개 이상의 샘플에 속한 항목을 아 형에 따라 Class 1, Class 2, 공통 발견으로 분류했다 (Fig. 6). Class 1에서는 세 포 분화 (GO:0060429, 변이된 유전자: BBS5, CD34, CTNNB1, ELF3,
ERRFI1, RYR2, SETD2, SLC12A2, SMAD4, SPRR2B, TBX18), Class 2에서
는 후성유전학적 조절과정 (GO:0006338, 변이된 유전자: BAZ2A, SUPT6H)에서 주로 변이가 발견되었다. 또한 두 아형에서 공통으로 세포 흡착 관련 유전자군 (GO:0007155, 변이된 유전자: CD34, COL6A5, CTNNB1, ENTPD1, ME1,
MOG, MUC4, MYBPC1, PCDHB11, PCDHB5, TRPM7)에서 변이가 발견되
Fig. 6. Heat map of gene ontology entries classified by gene expression classes.
H. 재현성 있는 변이의 검증
144개의 유전자 중 2개 이상의 샘플에서 발견된 유전자는 HMCN1과
UNC80이었다. HMCN1은 immunoglobulin superfamily에 속하며 세포 외 영역
에서 발견되고, UNC80은 sodium ion channel을 이루는 단백질이다. HMCN1이 AJHCC03과 AJHCC09 샘플에서, UNC80은 AJHCC02와 AJHCC12 샘플에서 발 견되었다. Sanger sequencing 검증을 실시한 결과 해당 염기 위치에서 heterozygous peak가 발견되어, 두 종류의 염기 서열이 공존함이 확인되었다. NGS 분석의 결과도 함께 IGV 소프트웨어를 이용해 도시되었다 (Table 5).
Table 5. IGV and Sanger sequencing validation of recurrent mutations.
ID Gene Sample Position Base Sequencing Validation
1 HMCN1 AJHCC03 AJHCC09 chr1:185,931,689 chr1:185,931,778 T>C G>A 2 UNC80 AJHCC12 AJHCC02 chr2:210,685,150 chr2:210,707,151 A>T G>C
I. 유전 변이체의 후성유전 결합위치와의 상관성 분석
엑솜 염기서열 분석에서 찾아낸 암 특이적 변이 중 후성유전 결합위치와 상 관성이 있는 생물학적 기능을 찾기 위해 HepG2 세포주의 후성유전 결합위치를 조사한 ChIP-seq 자료를 사용했다. 12 종류의 후성유전 변화 중 2 종류에서 Class 1과 2에 특이적인 module과 공통으로 나타나는 module이 발견되었다. 히 스톤 H3k09 trimethylation의 경우 12개 샘플에서 공통으로 RNA 전사 조절 (GO:0006350, 변이된 유전자: ARNT, ASXL3, BAZ2A, CCNT1, CHEK2, CNOT1, CTNNB1, ELF3, ERN1, IRF2, MNDA, MYSM1, NCOA6, SETD2,
SMAD4, SUPT6H, TBX18, ZNF175, ZNF239, ZNF845, ZNF883), 세포 흡착
(GO:0016337, 변이된 유전자: CD34, CTNNB1, ME1, PCDHB11, PCDHB5) 등에서 서열 변이가 발견되었고 (Fig. 5), 히스톤 H3k04 dimethylation의 경우 공 통으로는 noncoding RNA 처리 (GO:0034660, 변이된 유전자: DROSHA, IARS,
POP1, UTP20), RNA 대사 (GO:0006396, 변이된 유전자: CNOT1, DROSHA,
ERN1, POP1, RNASEL, SF3B1, UTP20) 등 후성유전학적 조절과 관련 있는
항목에서 서열 변이가 관찰되었다. Class 특이적인 항목의 경우 2개의 샘플에서 만 발견되어 통계적 의미는 없지만, Class 1에서는 RNA 전사 조절 (GO:0000122, 변이된 유전자: CTNNB1, IRF2, SMAD4, TBX18) (Fig. 7), Class 2에서는 ion transport (GO:43270, 변이된 유전자: SCN5A) 기능에서 서열 변이가 발견되었다 (Fig. 8).
Fig. 7. Heat map of gene ontology terms in the region labeled with histone H3k09me3. Gene ontology terms in G-protein coupled receptor signaling pathway, cell adhesion and regulation of transcription were enriched.
Fig. 8. Heat map of gene ontology terms in the region labeled with histone H4k04me2. Gene ontology terms in noncoding RNA metabolism, cell cycle, and cell differentiation were enriched in both Class 1 and Class 2. Class 1 showed enrichment in metabolism and regulation of transcription,
IV. 고 찰
다양한 진행 단계에서 얻은 간암 샘플에 대하여 엑솜 염기서열 분석과 유전 자 발현 분석을 실시하여 통합 분석했다. 유전자 발현 연구를 통해서는 암의 침 윤성에 따라 구분되는 2가지 아형을 발굴했다. Class 1에서는 물질 대사, Class 2 에서는 후성유전학적 조절과 세포 주기 등에 관여하는 유전자군의 발현양이 높 게 나타났다. 한편 임상 데이터에서 Class 2는 Class 1보다 크기가 더 작았고, 다 중 병변이 더 많이 발견되었으며, TNM stage T3, T4로 분류되었다. 따라서 두 아형은 암의 침윤성에 따라 발현 양상의 차이를 보임을 알 수 있다. 염기서열 분석을 통해서는 131,947개의 서열변이 집합을 얻었고, 다양한 기 준에 따라 신뢰성이 낮은 변이를 제거하여 149개의 서열변이를 구했다. 암 특이 적인 체세포 변이 중 driver 변이는 종양 발달의 원인이 되며, 암세포에 성장 속 도의 이점을 부여한다. 이들은 엑손 영역에 위치하여 단백질 서열에 변화를 주 며, 또한 여러 세대를 거치면서 변이가 있는 세포가 선호되므로, driver 변이는 무작위로 분포하지 않고 암 관련 유전자에서 발견될 확률이 높다. (Stratton 등, 2009; Wong 등, 2011). 따라서 단백질 암호화 영역에 위치하며 서열 변화를 일으 키는 변이는 저자의 filtering 방법을 통과했을 것으로 예측된다. 여러 환자에게서 공통으로 발견된 유전자의 변이는 HMCN1과 UNC80으로 총 2개가 발견되었다. Hemicentin 1 (HMCN1)은 세포 외부에 돌출된 immunoglobulin의 일종이며, 눈 질환인 황반 변성 (macular degeneration)과의 관계가 알려져 있다 (Brion 등, 2011). UNC80은 sodium leak cation channel (NALCN)을 구성하는 요소이며, G-protein coupled receptor 신호 전달에서 Src kinase를 고정하는 scaffold 단백질이다 (Ren, 2011; Lu와 Feng, 2012). HMCN1의 체세포 변이는 간암의 이전 연구 (Fujimoto 등, 2012; Guichard 등, 2012; Huang 등, 2012)에서 발견되었으며, 또한 전립선암 (Barbieri 등, 2012)과 직장암
(Cancer Genome Atlas Network, 2012)에서 발견되었다. UNC80의 변이는 간암 에서는 보고되지 않았으나, 직장암 (Cancer Genome Atlas Network, 2012)과 유 방암 (Stephens 등, 2012) 연구에서 발견되었다. 변이체의 대부분은 passenger 변이이므로, 나열된 유전자의 비교만으로는 변 이된 유전자가 담당한 주요 기능을 찾기 어렵다. 이러한 주요 기능은 찾기 위해 각 유전자 목록에 대한 gene ontology를 구하여 기능 분석을 시행했다. 기능 분 석에서 Class에 따라 다르게 변이된 유전자군과 전체에서 공통으로 변이된 유전 자군이 관찰되었다. 공통인 유전자군에서는 세포 흡착에서 높은 enrichment를 보 였으며, Class 1에서는 주로 세포 분화, Class 2에서는 후성유전학적 조절에 관한 유전자의 변이가 발견되었다. 유전자 영역에서와 다르게 gene ontology 항목간의 비교에서는 공통되는 기 능이 발견되었다. 이는 같은 유전자에서 변이가 일어나지 않더라도 같은 기능을 하는 집단에 속하는 유전자에 변이가 일어난다면 같은 양상의 변화가 일어날 수 있음을 암시한다. 특히, 양 Class의 샘플에서 공통으로 세포 흡착 관련 유전자의 변이가 관찰되었다. 세포 흡착 관련 유전자는 세포의 극성과 조직 내 결합력을 조절하며, 암세포는 흡착력이 정상 세포보다 떨어진다 (Hirohashi와 Kanai, 2003). 따라서 조사한 샘플에서도 각각 종류는 다르지만 세포 흡착 관련 유전자 에 변이가 일어났고, 이것이 암 형성에 관여하였음을 알 수 있다. Class 1에서는 세포 분화와 관련된 유전자군의 변이가 발견되었다. 증식상의 이점을 주는 암 유발 유전자에만 변이를 일으키면 immortalization이 일어나지 않으므로, 세포 분화 관련 유전자는 tumor-initiating gene으로 불리기도 한다 (Prasad 등, 2001). 간암 역시 cancer stem cell이 존재하는 것으로 생각되며, β -catenin의 변이와 간암 간의 높은 상관성을 볼 때 간암 세포의 stemness는 중 요한 것으로 생각된다 (Chiba 등, 2009).
NGS 연구를 통해 ARID2나 ARID1A와 같은 chromatin을 조절하는 유전자에서 기능 상실성 변이가 발견되었고, 전이성이 높은 HCC-LM6 세포주에서 ARID1A 의 서열 변이가 다수 관찰된 바 있다. 또한 Class 2에서 과발현된 유전자 기능 중에도 후성유전 조절이 포함되었다. 따라서 Class 2에서 집중되어 발견된 chromatin remodeling 관련 유전자의 변이는 이들의 높은 침윤성과 연관이 있을 것이다. HepG2 세포주의 ChIP-seq 결과를 이용한 후성유전 결합위치와의 상관성 분 석에서는 12 종류 중 2 종류에서 module이 발견되었다. 히스톤 H3k09 trimethylation (me3)에서 공통 module이, 히스톤 H3k04 dimethylation (me2)에 서 Class별 module이 발견되었다. 공통 module에서는 G-protein coupled receptor signaling과 RNA 전사 조절, 세포 흡착 등의 기능에서 변이가 발견되었 고, H3k09me3은 전사 억제 기능을 함이 알려져 있다 (Jenuwein과 Allis, 2011). 급성 골수성 백혈병 (acute myeloid leukemia, AML) 환자에 대한 H3k09me3 결 합 연구에서는 ETS, CRE와 같은 전사인자의 결합위치에서 H3k09me3 수준이 더 낮음이 발표되었다 (Muller-Tidow 등, 2010). 따라서 간암에서 H3k09me3 수 준이 높고 서열 변이가 일어난 유전자군은 ETS나 CRE 전사인자와 관련이 없을 것이며, 이들은 암 억제 유전자일 것으로 생각된다.
높은 히스톤 H3k04me2 수준과 서열 변이가 함께 발견된 유전자군에서는 세 포 주기, 단백질 ubiquitination, noncoding RNA 대사, 세포 분화 등의 기능이 공 통적으로 변이가 나타났다. 이 중 단백질의 ubiquitination 관련 유전자의 변이는 췌장암의 엑솜 염기서열 분석 연구에서 중복되어 발견된 바 있으며 (Wu 등, 2011), 종양 형성과 연관성이 높다. 각 Class별로 국한되어 변이된 기능은 공통 발견된 샘플의 수가 적어 통계적 의미는 없지만, Class 1에서는 물질 대사와 RNA 전사 조절, Class 2에서는 ion transport 기능에서 변이가 발견되었고, 이러 한 양상의 차이가 종양 악성화에 기여할 것으로 생각된다.
Class에서 공통으로 변이된 유전자군과 차이를 보인 유전자군을 밝힐 수 있었고, 아형별 비교 분석을 통해 간암의 악성화에 기여하는 서열 변이가 담당한 기능을 찾을 수 있었다. 향후 통합 분석의 범위를 ChIP-seq 등으로 넓혀 이질성이 높은 간암의 서열 변이체에서 핵심이 되는 과정을 찾는 데 기여할 것이다.
V. 결 론
이질성이 높은 간암 간의 차이를 규명하고 공통으로 변이된 기능을 발굴하 기 위해 유전자 발현과 서열 변이 영역의 통합 분석을 실시한 결과 다음과 같은 결론을 얻었다.
1. TNM staging system에 따른 T2와 T3/T4에 각각 대응하는 아형인 Class 1 과 Class 2가 유전자 발현 분석에서 발견되었으며, 이들은 간암의 침윤성 관련 유전자에서 발현상 차이를 보였다. 2. 여러 샘플에서 공통으로 변이가 발견된 유전자는 소수였지만, 세포 흡착, 세포 분화, 후성유전학적 조절 등의 기능과 연관된 유전자가 공통으로 변이되었다. 3. Class 1에서는 세포 분화 관련, Class 2에서는 후성유전 기능 조절 관련 유전 자의 변이가 공통으로 나타났고, Class 2의 높은 침윤성과 연관이 있을 것으로 생각된다. 4. 후성유전 결합위치와 변이체의 연관성 분석에서 전사 억제성 결합인 H3k09me3에서는 세포 신호전달, RNA 전사, 세포 흡착에서 공통된 module의 변 이가, 전사 활성화 결합인 H3k04me2에서는 세포 주기, 단백질 ubiquitination, noncoding RNA 대사, 세포 분화 등의 기능에서 변이가 관찰되었다.
변이체의 gene ontology 분석을 통해 각 Class에서 공통으로 변이된 유전자 군과 차이를 보인 유전자군을 밝힐 수 있었다.
참고문헌
1. Cancer Genome Atlas Network: Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330-337, 2012
2. American Cancer Society: Cancer Facts And Figures 2012. 2012
3. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, Auclair D, Onofrio RC, Park K, Kitabayashi N, MacDonald TY, Sheikh K, Vuong T, Guiducci C, Cibulskis K, Sivachenko A, Carter SL, Saksena G, Voet D, Hussain WM, Ramos AH, Winckler W, Redman MC, Ardlie K, Tewari AK, Mosquera JM, Rupp N, Wild PJ, Moch H, Morrissey C, Nelson PS, Kantoff PW, Gabriel SB, Golub TR, Meyerson M, Lander ES, Getz G, Rubin MA, Garraway LA: Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 44: 685-689, 2012
4. Bressac B, Kew M, Wands J, Ozturk M: Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 350: 429-431, 1991
5. Brion M, Sanchez-Salorio M, Corton M, de la Fuente M, Pazos B, Othman M, Swaroop A, Abecasis G, Sobrino B, Carracedo A: Genetic association study of age-related macular degeneration in the Spanish
population. Acta Ophthalmol 89: e12-22, 2011
6. Chaubert P, Gayer R, Zimmermann A, Fontolliet C, Stamm B, Bosman F, Shaw P: Germ-line mutations of the p16INK4(MTS1) gene occur in a subset of patients with hepatocellular carcinoma. Hepatology 25: 1376-1381, 1997
7. Chiba T, Kamiya A, Yokosuka O, Iwama A: Cancer stem cells in hepatocellular carcinoma: Recent progress and perspective. Cancer Lett 286: 145-153, 2009
8. de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, Perret C: Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A 95: 8847-8851, 1998
9. De Souza AT, Hankins GR, Washington MK, Orton TC, Jirtle RL: M6P/IGF2R gene is mutated in human hepatocellular carcinomas with loss of heterozygosity. Nat Genet 11: 447-449, 1995
10. Debuire B, Lemoine A: Liver: Hepatocellular carcinoma. Atlas Genet Cytogenet Oncol Haematol, 2001
11. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ: A framework for variation discovery and
genotyping using next-generation DNA sequencing data. Nat Genet 43: 491-498, 2011
12. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, Futreal PA: COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39: D945-950, 2011
13. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, Arai Y, Takahashi H, Shirakihara T, Nagasaki M, Shibuya T, Nakano K, Watanabe-Makino K, Tanaka H, Nakamura H, Kusuda J, Ojima H, Shimada K, Okusaka T, Ueno M, Shigekawa Y, Kawakami Y, Arihiro K, Ohdan H, Gotoh K, Ishikawa O, Ariizumi S, Yamamoto M, Yamada T, Chayama K, Kosuge T, Yamaue H, Kamatani N, Miyano S, Nakagama H, Nakamura Y, Tsunoda T, Shibata T, Nakagawa H: Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet 44: 760-764, 2012
14. Furukawa T, Kuboki Y, Tanji E, Yoshida S, Hatori T, Yamamoto M, Shibata N, Shimizu K, Kamatani N, Shiratori K: Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep 1: 161, 2011
Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clement B, Balabaud C, Chevet E, Laurent A, Couchy G, Letouze E, Calvo F, Zucman-Rossi J: Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet 44: 694-698, 2012
16. Hirohashi S, Kanai Y: Cell adhesion system and human cancer morphogenesis. Cancer Sci 94: 575-581, 2003
17. Ho DW, Yang ZF, Yi K, Lam CT, Ng MN, Yu WC, Lau J, Wan T, Wang X, Yan Z, Liu H, Zhang Y, Fan ST: Gene expression profiling of liver cancer stem cells by RNA-sequencing. PLoS One 7: e37159, 2012
18. Hou Y, Song L, Zhu P, Zhang B, Tao Y, Xu X, Li F, Wu K, Liang J, Shao D, Wu H, Ye X, Ye C, Wu R, Jian M, Chen Y, Xie W, Zhang R, Chen L, Liu X, Yao X, Zheng H, Yu C, Li Q, Gong Z, Mao M, Yang X, Yang L, Li J, Wang W, Lu Z, Gu N, Laurie G, Bolund L, Kristiansen K, Wang J, Yang H, Li Y, Zhang X: Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 148: 873-885, 2012
19. Huang da W, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44-57, 2009
Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet 44: 1117-1121, 2012
21. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D: Global cancer statistics. CA Cancer J Clin 61: 69-90, 2011
22. Jenuwein T, Allis CD: Translating the histone code. Science 293: 1074-1080, 2001
23. Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S, Diao J, Yeung S, Jubb A, Ye W, Wu TD, Kapadia SB, de Sauvage FJ, Gentleman RC, Stern HM, Seshagiri S, Pant KP, Modrusan Z, Ballinger DG, Zhang Z: The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res 22: 593-601, 2012
24. Jiao X, Wood LD, Lindman M, Jones S, Buckhaults P, Polyak K, Sukumar S, Carter H, Kim D, Karchin R, Sjoblom T: Somatic mutations in the Notch, NF-KB, PIK3CA, and Hedgehog pathways in human breast cancers. Genes Chromosomes Cancer 51: 480-489, 2012
25. Kawate S, Takenoshita S, Ohwada S, Mogi A, Fukusato T, Makita F, Kuwano H, Morishita Y: Mutation analysis of transforming growth factor beta type II receptor, Smad2, and Smad4 in hepatocellular carcinoma. Int J Oncol 14: 127-131, 1999
26. Kita R, Nishida N, Fukuda Y, Azechi H, Matsuoka Y, Komeda T, Sando T, Nakao K, Ishizaki K: Infrequent alterations of the p16INK4A
gene in liver cancer. Int J Cancer 67: 176-180, 1996
27. Li H: Improving SNP discovery by base alignment quality. Bioinformatics 27: 1157-1158, 2011
28. Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754-1760, 2009
29. Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA, Pawlik TM, Daniel HD, Kannangai R, Offerhaus GJ, Velculescu VE, Wang L, Zhou S, Vogelstein B, Hruban RH, Papadopoulos N, Cai J, Torbenson MS, Kinzler KW: Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet 43: 828-829, 2011
30. Lu TZ, Feng ZP: NALCN: a regulator of pacemaker activity. Mol Neurobiol 45: 415-423, 2012
31. Matsuzaki K, Murata M, Yoshida K, Sekimoto G, Uemura Y, Sakaida N, Kaibori M, Kamiyama Y, Nishizawa M, Fujisawa J, Okazaki K, Seki T: Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 46: 48-57, 2007
32. McGivern DR, Lemon SM: Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene 30: 1969-1983, 2011
Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297-1303, 2010
34. Michielsen P, Ho E: Viral hepatitis B and hepatocellular carcinoma. Acta Gastroenterol Belg 74: 4-8, 2011
35. Miyoshi Y, Iwao K, Nagasawa Y, Aihara T, Sasaki Y, Imaoka S, Murata M, Shimano T, Nakamura Y: Activation of the beta-catenin gene in primary hepatocellular carcinomas by somatic alterations involving exon 3. Cancer Res 58: 2524-2527, 1998
36. Muller-Tidow C, Klein HU, Hascher A, Isken F, Tickenbrock L, Thoennissen N, Agrawal-Singh S, Tschanter P, Disselhoff C, Wang Y, Becker A, Thiede C, Ehninger G, zur Stadt U, Koschmieder S, Seidl M, Muller FU, Schmitz W, Schlenke P, McClelland M, Berdel WE, Dugas M, Serve H: Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood 116: 3564-3571, 2010
37. Murakami Y, Hayashi K, Hirohashi S, Sekiya T: Aberrations of the tumor suppressor p53 and retinoblastoma genes in human hepatocellular carcinomas. Cancer Res 51: 5520-5525, 1991
38. Nakamura T, Iwamura Y, Kaneko M, Nakagawa K, Kawai K, Mitamura K, Futagawa T, Hayashi H: Deletions and rearrangements
some neurogenic tumors as found in a study of 121 tumors. Jpn J Clin Oncol 21: 325-329, 1991
39. Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ: Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42: 30-35, 2010
40. Nishida N, Fukuda Y, Komeda T, Kita R, Sando T, Furukawa M, Amenomori M, Shibagaki I, Nakao K, Ikenaga M, et al.: Amplification and overexpression of the cyclin D1 gene in aggressive human hepatocellular carcinoma. Cancer Res 54: 3107-3110, 1994
41. Prasad KN, Hovland AR, Nahreini P, Cole WC, Hovland P, Kumar B, Prasad KC: Differentiation genes: are they primary targets for human carcinogenesis? Exp Biol Med (Maywood) 226: 805-813, 2001
42. Puda A, Milosevic JD, Berg T, Klampfl T, Harutyunyan AS, Gisslinger B, Rumi E, Pietra D, Malcovati L, Elena C, Doubek M, Steurer M, Tosic N, Pavlovic S, Guglielmelli P, Pieri L, Vannucchi AM, Gisslinger H, Cazzola M, Kralovics R: Frequent deletions of JARID2 in leukemic transformation of chronic myeloid malignancies. Am J Hematol 87: 245-250, 2012
43. Puisieux A, Ozturk M: TP53 and hepatocellular carcinoma. Pathol Biol (Paris) 45: 864-870, 1997
carcinoma. World J Gastroenterol 8: 385-392, 2002
45. Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, Ramsay AJ, Bea S, Pinyol M, Martinez-Trillos A, Lopez-Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, Delgado J, Gine E, Hernandez JM, Gonzalez-Diaz M, Puente DA, Velasco G, Freije JM, Tubio JM, Royo R, Gelpi JL, Orozco M, Pisano DG, Zamora J, Vazquez M, Valencia A, Himmelbauer H, Bayes M, Heath S, Gut M, Gut I, Estivill X, Lopez-Guillermo A, Puente XS, Campo E, Lopez-Otin C: Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 44: 47-52, 2012
46. Raney BJ, Cline MS, Rosenbloom KR, Dreszer TR, Learned K, Barber GP, Meyer LR, Sloan CA, Malladi VS, Roskin KM, Suh BB, Hinrichs AS, Clawson H, Zweig AS, Kirkup V, Fujita PA, Rhead B, Smith KE, Pohl A, Kuhn RM, Karolchik D, Haussler D, Kent WJ: ENCODE whole-genome data in the UCSC genome browser (2011 update). Nucleic Acids Res 39: D871-875, 2011
47. Ren D: Sodium leak channels in neuronal excitability and rhythmic behaviors. Neuron 72: 899-911, 2011
48. Roth A, Ding J, Morin R, Crisan A, Ha G, Giuliany R, Bashashati A, Hirst M, Turashvili G, Oloumi A, Marra MA, Aparicio S, Shah SP: JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data.
Bioinformatics 28: 907-913, 2012
49. Saldanha AJ: Java Treeview--extensible visualization of microarray data. Bioinformatics 20: 3246-3248, 2004
50. Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, Sasaki Y, Imaoka S, Murata M, Shimano T, Yamaoka Y, Nakamura Y: AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet 24: 245-250, 2000
51. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J, Easton D, Langerod A, Lee MT, Shen CY, Tee BT, Huimin BW, Broeks A, Vargas AC, Turashvili G, Martens J, Fatima A, Miron P, Chin SF, Thomas G, Boyault S, Mariani O, Lakhani SR, van de Vijver M, van 't Veer L, Foekens J, Desmedt C, Sotiriou C, Tutt A, Caldas C, Reis-Filho JS, Aparicio SA, Salomon AV, Borresen-Dale AL, Richardson AL, Campbell PJ, Futreal PA, Stratton MR: The landscape of cancer genes and mutational processes in breast cancer. Nature 486: 400-404, 2012
52. Stratton MR, Campbell PJ, Futreal PA: The cancer genome. Nature 458: 719-724, 2009
53. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP: Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545-15550, 2005
54. Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, Mulawadi FH, Wong KF, Liu AM, Poon RT, Fan ST, Chan KL, Gong Z, Hu Y, Lin Z, Wang G, Zhang Q, Barber TD, Chou WC, Aggarwal A, Hao K, Zhou W, Zhang C, Hardwick J, Buser C, Xu J, Kan Z, Dai H, Mao M, Reinhard C, Wang J, Luk JM: Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet 44: 765-769, 2012
55. Suzuki R, Shimodaira H: Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22: 1540-1542, 2006
56. Tao Y, Ruan J, Yeh SH, Lu X, Wang Y, Zhai W, Cai J, Ling S, Gong Q, Chong Z, Qu Z, Li Q, Liu J, Yang J, Zheng C, Zeng C, Wang HY, Zhang J, Wang SH, Hao L, Dong L, Li W, Sun M, Zou W, Yu C, Li C, Liu G, Jiang L, Xu J, Huang H, Mi S, Zhang B, Chen B, Zhao W, Hu S, Zhuang SM, Shen Y, Shi S, Brown C, White KP, Chen DS, Chen PJ, Wu CI: Rapid growth of a hepatocellular carcinoma and the driving mutations revealed by cell-population genetic analysis of whole-genome data. Proc Natl Acad Sci U S A 108: 12042-12047, 2011
57. Thorvaldsdottir H, Robinson JT, Mesirov JP: Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform, 2012
58. Totoki Y, Tatsuno K, Yamamoto S, Arai Y, Hosoda F, Ishikawa S, Tsutsumi S, Sonoda K, Totsuka H, Shirakihara T, Sakamoto H, Wang L, Ojima H, Shimada K, Kosuge T, Okusaka T, Kato K, Kusuda J, Yoshida T, Aburatani H, Shibata T: High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet 43: 464-469, 2011
59. Wang K, Li M, Hakonarson H: ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164, 2010
60. Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, Zhang W, Vartanov AR, Fernandes SM, Goldstein NR, Folco EG, Cibulskis K, Tesar B, Sievers QL, Shefler E, Gabriel S, Hacohen N, Reed R, Meyerson M, Golub TR, Lander ES, Neuberg D, Brown JR, Getz G, Wu CJ: SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 365: 2497-2506, 2011
61. Wang L, Tsutsumi S, Kawaguchi T, Nagasaki K, Tatsuno K, Yamamoto S, Sang F, Sonoda K, Sugawara M, Saiura A, Hirono S, Yamaue H, Miki Y, Isomura M, Totoki Y, Nagae G, Isagawa T, Ueda H, Murayama-Hosokawa S, Shibata T, Sakamoto H, Kanai Y, Kaneda
A, Noda T, Aburatani H: Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res 22: 208-219, 2012
62. Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, Gomes X, Tartaro K, Niazi F, Turcotte CL, Irzyk GP, Lupski JR, Chinault C, Song XZ, Liu Y, Yuan Y, Nazareth L, Qin X, Muzny DM, Margulies M, Weinstock GM, Gibbs RA, Rothberg JM: The complete genome of an individual by massively parallel DNA sequencing. Nature 452: 872-876, 2008
63. Wong KM, Hudson TJ, McPherson JD: Unraveling the genetics of cancer: genome sequencing and beyond. Annu Rev Genomics Hum Genet 12: 407-430, 2011
64. Wu J, Jiao Y, Dal Molin M, Maitra A, de Wilde RF, Wood LD, Eshleman JR, Goggins MG, Wolfgang CL, Canto MI, Schulick RD, Edil BH, Choti MA, Adsay V, Klimstra DS, Offerhaus GJ, Klein AP, Kopelovich L, Carter H, Karchin R, Allen PJ, Schmidt CM, Naito Y, Diaz LA, Jr., Kinzler KW, Papadopoulos N, Hruban RH, Vogelstein B: Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A 108: 21188-21193, 2011
65. Xu X, Hou Y, Yin X, Bao L, Tang A, Song L, Li F, Tsang S, Wu K, Wu H, He W, Zeng L, Xing M, Wu R, Jiang H, Liu X, Cao D,
Guo G, Hu X, Gui Y, Li Z, Xie W, Sun X, Shi M, Cai Z, Wang B, Zhong M, Li J, Lu Z, Gu N, Zhang X, Goodman L, Bolund L, Wang J, Yang H, Kristiansen K, Dean M, Li Y: Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell 148: 886-895, 2012
66. Yakicier MC, Irmak MB, Romano A, Kew M, Ozturk M: Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene 18: 4879-4883, 1999
67. Yamada T, De Souza AT, Finkelstein S, Jirtle RL: Loss of the gene encoding mannose 6-phosphate/insulin-like growth factor II receptor is an early event in liver carcinogenesis. Proc Natl Acad Sci U S A 94: 10351-10355, 1997
68. Ye SL, Takayama T, Geschwind J, Marrero JA, Bronowicki JP: Current approaches to the treatment of early hepatocellular carcinoma. Oncologist 15 Suppl 4: 34-41, 2010
69. Zhao H, Wang J, Han Y, Huang Z, Ying J, Bi X, Zhao J, Fang Y, Zhou H, Zhou J, Li Z, Zhang Y, Yang X, Yan T, Wang L, Torbenson MS, Cai J: ARID2: a new tumor suppressor gene in hepatocellular carcinoma. Oncotarget 2: 886-891, 2011
70. Zhou X, Ren L, Meng Q, Li Y, Yu Y, Yu J: The next-generation sequencing technology and application. Protein Cell 1: 520-536, 2010
ID Sample Gene Amino Acid Position Change References 1 AJHCC06 MORN1 NM_024848:c.G316A:p.G106S chr1:2318900-2318900 C>T Guichard 2012 2 AJHCC02 ERRFI1 NM_018948:c.281delC:p.P94fs chr1:8074378-8074378 GΔ Fujimoto 2012 3 AJHCC08 MAN1C1 NM_020379:c.G1147A:p.V383I chr1:26098153-26098153 G>A 4 AJHCC03 MYSM1 NM_001085487:c.A2250T:p.E750D chr1:59127098-59127098 T>A 5 AJHCC09 CLCA1 NM_001285:c.T1829C:p.V610A chr1:86960018-86960018 T>C 6 AJHCC02 PALMD NM_017734:c.C955A:p.H319N chr1:100154771-100154771 C>A 7 AJHCC03 VPS45 NM_007259:c.C774G:p.D258E chr1:150053510-150053510 C>G 8 AJHCC09 ARNT NM_001197325:c.T713C:p.M238T chr1:150807059-150807059 A>G 9 AJHCC10 RPTN NM_001122965:c.C1033A:p.Q345K chr1:152128542-152128542 G>T 10 AJHCC06 SPRR2B NM_001017418:c.C59T:p.T20M chr1:153043257-153043257 G>A 11 AJHCC03 DAP3 NM_001199850:c.A541G:p.S181G chr1:155698872-155698872 A>G 12 AJHCC07 MNDA NM_002432:c.A1159C:p.S387R chr1:158817689-158817689 A>C Li 2011 13 AJHCC07 MNDA NM_002432:c.T1168G:p.F390V chr1:158817698-158817698 T>G Li 2011 14 AJHCC07 TOMM40L NM_032174:c.A892T:p.R298X chr1:161198850-161198850 A>T Guichard 2012 15 AJHCC09 NME7 NM_013330:c.A507G:p.I169M chr1:169267935-169267935 T>C 16 AJHCC09 STX6 NM_005819:c.C640T:p.R214W chr1:180953864-180953864 G>A 17 AJHCC09 CACNA1E NM_001205294:c.G5027A:p.G1676D chr1:181741312-181741312 G>A 18 AJHCC06 RNASEL NM_021133:c.T320C:p.L107P chr1:182555622-182555622 A>G 19 AJHCC03 HMCN1 NM_031935:c.T1868C:p.F623S chr1:185931689-185931689 T>C Guichard2012;
Fujimoto 2012 20 AJHCC09 HMCN1 NM_031935:c.G1957A:p.V653M chr1:185931778-185931778 G>A Guichard2012;
Fujimoto 2012 21 AJHCC10 ELF3 NM_001114309:c.G1064A:p.G355D chr1:201984399-201984399 G>A 22 AJHCC11 CD34 NM_001025109:c.G236A:p.G79D chr1:208073192-208073192 C>T 23 AJHCC09 RYR2 NM_001035:c.C10699T:p.R3567C chr1:237889582-237889582 C>T Guichard 2012 24 AJHCC04 C1orf101 NM_001242340:c.C1166T:p.A389V chr1:244735743 C>T
부 록
25 AJHCC04 THUMPD2 NM_025264:c.G586C:p.A196P chr2:39996936-39996936 C>G 26 AJHCC09 CCDC88A NM_001135597:c.G1606C:p.A536P chr2:55563867-55563867 C>G 27 AJHCC05 LRP1B NM_018557:c.G13071T:p.E4357D chr2:141032064-141032064 C>A
Li 2011; Guichard 2012; Fujimoto 2012 28 AJHCC03 XIRP2 NM_001199144:c.A8371G:p.T2791A chr2:168106939-168106939 A>G Fujimoto 2012 29 AJHCC11 BBS5 NM_152384:c.G66A:p.M22I chr2:170338767-170338767 G>A 30 AJHCC07 TMEM194B NM_001142645:c.A595G:p.K199E chr2:191382263-191382263 T>C 31 AJHCC03 SF3B1 NM_012433:c.A2221C:p.K741Q chr2:198266711-198266711 T>G 32 AJHCC09 AOX1 NM_001159:c.G3826T:p.G1276W chr2:201534325-201534325 G>T 33 AJHCC03 MDH1B NM_001039845:c.C787G:p.L263V chr2:207619856-207619856 G>C 34 AJHCC12 UNC80 NM_032504:c.A2078T:p.E693V chr2:210685150-210685150 A>T 35 AJHCC02 UNC80 NM_032504:c.G3441C:p.E1147D chr2:210707151-210707151 G>C 36 AJHCC03 USP40 NM_018218:c.C1516T:p.P506S chr2:234438147-234438147 G>A 37 AJHCC03 SPP2 NM_006944:c.G364A:p.V122I chr2:234969043-234969043 G>A 38 AJHCC03 ANKRD28 NM_001195099:c.G2252T:p.S751I chr3:15718550-15718550 C>A 39 AJHCC07 GOLGA4 NM_002078:c.A3526G:p.S1176G chr3:37366903-37366903 A>G Guichard 2012 40 AJHCC02 EXOG NM_005107:c.G164T:p.G55V chr3:38539120-38539120 G>T 41 AJHCC09 SCN5A NM_001099405:c.G5005A:p.G1669S chr3:38592804-38592804 C>T 42 AJHCC02 CTNNB1 NM_001098209:c.T1161G:p.N387K chr3:41274911-41274911 T>G
Li 2011; Guichard 2012; Fujimoto 2012 43 AJHCC02 C3orf23 NM_173826:c.A1022G:p.Y341C chr3:44441983-44441983 A>G 44 AJHCC10 SETD2 NM_014159:c.G5340A:p.W1780X chr3:47127742-47127742 C>T 45 AJHCC09 CACNA2D3 NM_018398:c.T2666A:p.L889X chr3:55021756-55021756 T>A 46 AJHCC09 OR5AC2 NM_054106:c.C166A:p.H56N chr3:97806182-97806182 C>A 47 AJHCC09 SIDT1 NM_017699:c.T994A:p.Y332N chr3:113304110-113304110 T>A Li 2011 48 AJHCC03 COL6A5 NM_153264:c.T4013A:p.V1338E chr3:130119896-130119896 T>A 49 AJHCC03 PCCB NM_000532:c.G470C:p.G157A chr3:135980834-135980834 G>C 50 AJHCC12 SI NM_001041:c.C3487T:p.H1163Y chr3:164735791-164735791 G>A 51 AJHCC09 GNB4 NM_021629:c.A154T:p.R52W chr3:179137236-179137236 T>A
-195516488
53 AJHCC03 TIGD2 NM_145715:c.A670T:p.N224Y chr4:90034795-90034795 A>T 54 AJHCC01 LRBA NM_001199282:c.C5456T:p.A1819V chr4:151727485-151727485 G>A 55 AJHCC10 IRF2 NM_002199:c.A119T:p.H40L chr4:185340691-185340691 T>A 56 AJHCC10 DROSHA NM_001100412:c.T3374C:p.L1125S chr5:31421419-31421419 A>G 57 AJHCC03 RNF180 NM_001113561:c.C971G:p.T324S chr5:63510124-63510124 C>G 58 AJHCC04 ANKRD32 NM_032290:c.C632T:p.S211F chr5:93985196-93985196 C>T 59 AJHCC09 SLC12A2 NM_001046:c.A1023G:p.I341M chr5:127450348-127450348 A>G 60 AJHCC08 C5orf48 NM_207408:c.237_238del:p.79_80del chr5:129571765-129571766 CTΔ 61 AJHCC07 MATR3 NM_001194956:c.G268T:p.A90S chr5:138652744-138652744 G>T 62 AJHCC03 PCDHB5 NM_015669:c.G955T:p.V319L chr5:140515971-140515971 G>T 63 AJHCC10 PCDHB11 NM_018931:c.C1157A:p.P386Q chr5:140580504-140580504 C>A 64 AJHCC09 G3BP1 NM_005754:c.A1186T:p.S396C chr5:151180422-151180422 A>T 65 AJHCC03 KIF4B NM_001099293:c.C2941T:p.Q981X chr5:154396360-154396360 C>T 66 AJHCC06 HIST1H2BD NM_021063:c.A287G:p.Q96R chr6:26158684-26158684 A>G 67 AJHCC07 HIST1H2AL NM_003511:c.G295C:p.G99R chr6:27833427-27833427 G>C 68 AJHCC01 OR2J 3 NM_001005216:c.A406T:p.T136S chr6:29080073-29080073 A>T 69 AJHCC09 MOG NM_001170418:c.A224C:p.E75A chr6:29635681-29635681 A>C 70 AJHCC10 MUC21 NM_001010909:c.G667A:p.G223S chr6:30954619-30954619 G>A 71 AJHCC02 SLC26A8 NM_138718:c.T2594G:p.V865G chr6:35911681-35911681 A>C 72 AJHCC06 C6orf132 NM_001164446:c.C2065A:p.P689T chr6:42073585-42073585 G>T 73 AJHCC09 GSTA4 NM_001512:c.A391G:p.R131G chr6:52849285-52849285 T>C 74 AJHCC03 ME1 NM_002395:c.C508T:p.P170S chr6:84055984-84055984 G>A 75 AJHCC08 TBX18 NM_001080508:c.T691G:p.S231A chr6:85466496-85466496 A>C 76 AJHCC01 C6orf186 NM_001123364:c.T949A:p.W317R chr6:110567301-110567301 A>T 77 AJHCC11 FYN NM_153047:c.T1184A:p.V395E chr6:112015658-112015658 A>T 78 AJHCC10 CEP85L NM_001042475:c.G1246A:p.A416T chr6:118832472-118832472 C>T 79 AJHCC11 RPS12 NM_001016:c.C301T:p.R101C chr6:133138165-133138165 C>T 80 AJHCC06 C6orf211 NM_024573:c.G975A:p.M325I chr6:151789894-151789894 G>A