www.cdc.go.kr
1178
들어가는 말
분자생물학에 있어 라이브러리란 유전자 클로닝을 통해서 각기 다른 DNA 단편 조각을 동일한 벡터에 삽입한 모음을 말한다. 라이브러리는 서로 다른 개체의 총 유전체 DNA를 제한효소로 잘라 만든 유전체 라이브러리(genomic library)와 암호화 서열만을 포함하고 있는 cDNA 라이브러리(cDNA library)가 있다. 이들은 특정 개체의 유전체 염기서열 분석에서 새로운 유전자의 발견과 기능연구까지 넓은 분야에 이용되고 있다[1]. 또한 유전물질의 염기서열을 분석하기 위한 생거 시퀀싱(Sanger sequencing)에도 라이브러리의 제작이 필수적이다[2]. 라이브러리의 제작은 1세대 시퀀싱부터 최근 3세대 시퀀싱까지 그 방법과 기작이 플랫폼에 따라 다르지만 질(quality)이 좋은 시퀀싱 데이터를 얻는 중요한 단계라는 것은 모든 플랫폼에서 공통적이다. 이 글에서는 2세대 시퀀싱(Next generation sequencing, NGS)부터 3세대 시퀀싱(3rd generation sequencing)까지 라이브러리 제작 방법과 병원체(박테리아,Library construction techniques for pathogen whole genome sequencing
Yang Hyo-Jin, Choi Hyun-Jung, Chai Hee-Youl, Kang Yeon-Ho* Division of Biosafety Evaluation and Control, KNIH, KCDC
In molecular biology, libraries are widely used for analyzing the sequences of gene or genome to discover a new gene and its function. Furthermore, libraries are essential in genomics for preparing genetic materials into a form compatible with the sequencing system to be used. For this reason, the library preparation method has evolved from that applied in first-generation sequencing techniques (Sanger sequencing) to more recent sequencing technology [second- first-generation sequencing i.e., next-generation sequencing (NGS) and third-generation sequencing]. Moreover, the methods are changed depending on the type of target genetic materials (e.g., DNA, RNA). In this report, we introduce general library preparation methods and specific library construction methods according to the type of pathogens being detected.
Keywords: Library construction, Pathogen, Whole genome sequencing, Genome, Genetic material
질병관리본부 국립보건연구원 생물안전평가과 양효진, 최현정, 채희열, 강연호* *교신저자: slowpc@korea.kr, 043-719-8040
병원체 염기서열 생산을 위한
www.cdc.go.kr
1179 바이러스)의 전장 유전체 시퀀싱(whole genome sequencing)에
적용하는 라이브러리의 제작 방법을 소개하고자 한다.
몸 말
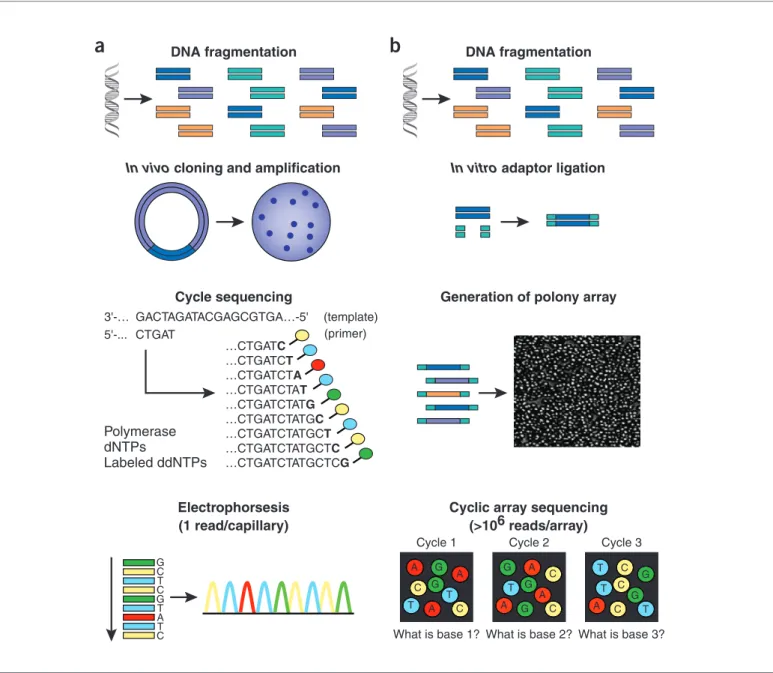
시퀀싱에 있어 라이브러리의 제작 과정은 각종 유전물질들(DNA, RNA)을 시퀀싱 시스템과 호환되도록 준비하는 과정이다. 1세대 시퀀싱은 미생물 내에 클로닝 벡터를 삽입해서 in vivo 상태에서 복제, 증폭(colony) 후 이들 서열을 분석했다면, 2세대 시퀀싱(Next generation sequencing, NGS)부터는 flow cell이나 bead와 같은 in vitro 상에서 증폭(polony) 하여 서열을 분석한다(Figure 1).라이브러리 제작 단계는 유전물질의 단편화(fragmentation), 어댑터 부착(adaptor ligation), 라이브러리의 정량 및 사이즈 확인 과정(quality control, QC)으로 완성된다(Figure 2). 첫 번째, 유전물질의 단편화 과정은 라이브러리 제작에 중요 지표로 각각의 플랫폼이 생산할 수 있는 리드(read)의 사이즈에 따라 단편의 길이를 결정할 수 있다. 2세대 시퀀싱에 비해서 3세대 시퀀싱 리드의
Molecule Sequencer technology (Helicos; Cambridge, MA, USA). The concept of cyclic-array sequencing can be summarized as the sequencing of a dense array of DNA features by iterative cycles of enzymatic manipu-lation and imaging-based data collection15 (Shendure and colleagues16). Two reports in 2005 described the first integrated implementations of cyclic-array strategies that were both practical and cost-competitive with conventional sequencing (J.S. et al.13 and ref. 14), and other groups have quickly followed17,18.
Although these platforms are quite diverse in sequencing biochem-istry as well as in how the array is generated, their work flows are conceptually similar (Fig. 1b). Library preparation is accomplished by random fragmentation of DNA, followed by in vitro ligation of common adaptor sequences. Alternative protocols can be used to generate jumping libraries of mate-paired tags with control-lable distance distributions13,19. The genera-tion of clonally clustered amplicons to serve as sequencing features can be achieved by several approaches, including in situ polo-nies15, emulsion PCR20 or bridge PCR21,22 (Fig. 2). What is common to these methods is that PCR amplicons derived from any given single library molecule end up spatially clus-tered, either to a single location on a planar substrate (in situ polonies, bridge PCR), or to the surface of micron-scale beads, which can be recovered and arrayed (emulsion PCR). The sequencing process itself consists of alternating cycles of enzyme-driven bio-chemistry and imaging-based data acquisi-tion (Fig. 3). The platforms that are discussed here all rely on sequencing by synthesis, that is, serial extension of primed templates, but the enzyme driving the synthesis can be either a polymerase16,23 or a ligase13,24. Data are acquired by imaging of the full array at each cycle (e.g., of fluorescently labeled nucleotides incorporated by a polymerase).
Global advantages of second-generation or cyclic-array strategies, relative to Sanger sequencing, include the following: (i) in vitro construction of a sequencing library, followed by in vitro clonal amplification to generate sequencing features, circumvents several bot-tlenecks that restrict the parallelism of con-ventional sequencing (that is, transformation of E. coli and colony picking). (ii) Array-based sequencing enables a much higher degree of parallelism than conventional capillary-based sequencing. As the effective size of sequencing features can be on the order of 1 µm, hundreds of millions of sequencing reads can potentially be obtained in parallel by rastered imaging of a reasonably sized surface area. (iii) Because array features are immobilized to a planar sur-face, they can be enzymatically manipulated by a single reagent volume. Although microliter-scale reagent volumes are used in practice, these are essentially amortized over the full set of sequencing features on the array, dropping Second-generation DNA sequencing
Alternative strategies for DNA sequencing can be grouped into several categories (as discussed previously in ref. 4). These include (i) microelec-trophoretic methods9 (Box 1), (ii) sequencing by hybridization10 (Box 2), (iii) real-time observation of single molecules11,12 (Box 3) and (iv) cyclic-array sequencing (J.S. et al.13 and ref. 14). Here, we use ‘second-generation’ in reference to the various implementations of cyclic-array sequencing that have recently been realized in a commercial product (e.g., 454 sequencing (used in the 454 Genome Sequencers, Roche Applied Science; Basel), Solexa technology (used in the Illumina (San Diego) Genome Analyzer), the SOLiD platform (Applied Biosystems; Foster City, CA, USA), the Polonator (Dover/Harvard) and the HeliScope Single
3'-… GACTAGATACGAGCGTGA…-5' (template) 5'-... CTGAT (primer) …CTGATC …CTGATCT …CTGATCTA …CTGATCTAT …CTGATCTATG …CTGATCTATGC …CTGATCTATGCT …CTGATCTATGCTC …CTGATCTATGCTCG A G G A A C T T C A G G A A C T T C G A G C G C A A T G A G C G C A A T T C C G C T G A T T C C G C T G A T Cyclic array sequencing
(>106 reads/array)
Cycle 1 Cycle 2 Cycle 3
What is base 1? What is base 2? What is base 3?
a
DNA fragmentationIn vivo cloning and amplification In vitro adaptor ligation
Electrophorsesis (1 read/capillary) Polymerase dNTPs Labeled ddNTPs G C T C G T A T C
b
DNA fragmentationCycle sequencing Generation of polony array
Figure 1 Work flow of conventional versus second-generation sequencing. (a) With high-throughput
shotgun Sanger sequencing, genomic DNA is fragmented, then cloned to a plasmid vector and used to transform E. coli. For each sequencing reaction, a single bacterial colony is picked and plasmid DNA isolated. Each cycle sequencing reaction takes place within a microliter-scale volume, generating a ladder of ddNTP-terminated, dye-labeled products, which are subjected to high-resolution electrophoretic separation within one of 96 or 384 capillaries in one run of a sequencing instrument. As fluorescently labeled fragments of discrete sizes pass a detector, the four-channel emission spectrum is used to generate a sequencing trace. (b) In shotgun sequencing with cyclic-array methods, common
adaptors are ligated to fragmented genomic DNA, which is then subjected to one of several protocols that results in an array of millions of spatially immobilized PCR colonies or ‘polonies’15. Each polony
consists of many copies of a single shotgun library fragment. As all polonies are tethered to a planar array, a single microliter-scale reagent volume (e.g., for primer hybridization and then for enzymatic extension reactions) can be applied to manipulate all array features in parallel. Similarly, imaging-based detection of fluorescent labels incorporated with each extension can be used to acquire sequencing
R E v I E W
© 200 8 Nature Pub lishing Gr oup http://www .nature .com/naturebiotec hnologywww.cdc.go.kr
1180 길이(PacBio Sequel의 경우 최대 40 Kb)가 길기 때문에 시료의
단편도 길게 제작된다. DNA 단편화 과정은 물리적인 방법(physical methods: acoustic shearing, sonication)과 효소법(enzymatic methods: non-specific endonuclease cocktails, transposase tagmentation)으로 나눈다. 두 방법 모두 유전물질을 단편화하기에 효율적이지만 효소법의 경우 물리적인 방법에 비해 인위적인 indel(염기 삽입과 결실) 부위를 더 많이 생성한다[3]. 두 번째, 어댑터 부착 과정은 단편화된 유전물질에 시퀀싱 시스템 내의 flow cell이나 bead에 부착된 올리고머에 상보적인 올리고머(어댑터)와 각각의 시료를 인식할 수 있는 인덱스를 부착하는 과정이다. 이 과정에서 PCR을 통해 라이브러리의 양을 증가시켜준다. 마지막으로 완성된 라이브러리의 사이즈를 확인하고 정량하는 과정이다. 이 과정은 완성된 라이브러리를 검증하는 과정으로 ‘라이브러리 품질관리(QC)’라고도 하고 시퀀싱의 질을 결정하는 단계이기도 하다. 이 과정에서는 라이브러리의 사이즈 분포와 정량이 매우 정확하게 측정되어야 한다. 라이브러리의 양이 많이 주입되는 경우 flow cell이나 bead에 라이브러리가 포화상태가 되어 시퀀스를 읽는데 문제가 발생하고, 라이브러리의 양이 적게 주입되는 경우 원하는 만큼의 시퀀싱 범위(coverage)와 시퀀싱 배수(read depth)를 얻을 수 없다. 따라서 라이브러리 정량은 double strand DNA만 특이적으로 정량하는 형광법(Qubit, PicoGreen)이나 어댑터가 부착된 라이브러리만 정량하는 q-PCR 방법을 사용한다[4].
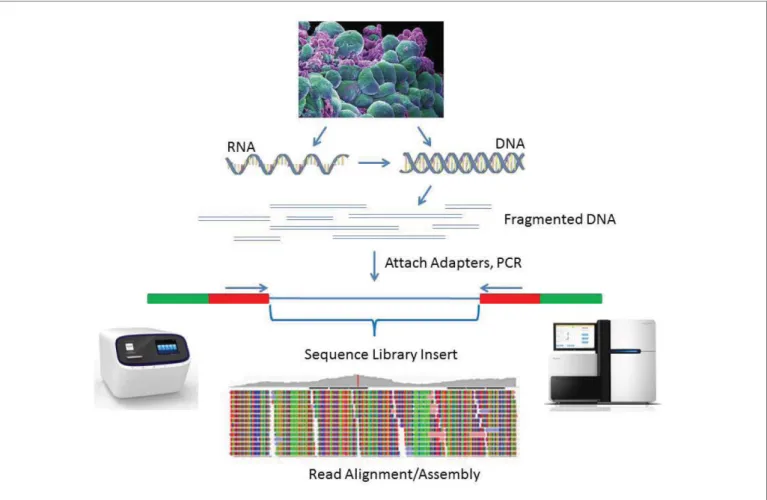
앞서 설명한 라이브러리 제작방법은 병원체의 전장 유전체 시퀀싱(whole genome sequencing)에도 적용이 된다. 박테리아는 바이러스에 비해 유전체의 사이즈도 크고(Mega base 단위) 질이 좋은 시료를 다량으로 준비하기 비교적 쉽다. 반면 바이러스의 경우 유전체의 종류도 다양하고(ssRNA, dsRNA, DNA) 유전체의 사이즈(Kilo base 단위)도 작아 라이브러리 제작에 알맞은 시료를
준비하기에 어려움이 많다.
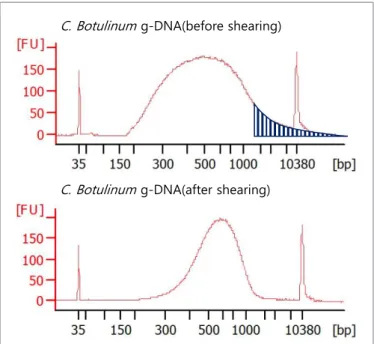
현재 본 원에서 다루는 박테리아 유전체를 이용한 라이브러리 제작은 Illumina사(San Diego, Califonia, USA)의 전용 키트를 사 용 하고 있다. 이 키 트는 효 소 를 이 용 해 서 유 전 물 질 을 단편화시키는 동시에 어댑터를 부착시키는 tagmentation 기술(Figure 3)로 시료를 다루기 쉽고 라이브러리 제작 시간을 단축시켜주지만, 저농도(0.2 ng/uL)를 사용하기 때문에 비교적 농도가 높은 박테리아 유전체는 여러 번 희석하는 과정을 거쳐야 하는 단점이 있다[3]. 또한, GC contents(%)가 특이적으로 높거나 낮을 때, 효소에 의한 라이브러리의 단편화가 완벽하지 않아 비교적 큰 사이즈의 라이브러리가 남아있는 경우가 있다. 이러한 경우엔 물리적인 방법으로 단편화시킨 후 효소를 이용한 방법을 적용시킨다(Figure 4). 바이러스 유전체는 사이즈가 작고 유전물질의 바이러스 마다 다르기 때문에 고유 형태를 그대로 사용하지 않고 유전체가 끊어지지 않도록 연결된 PCR 산물이나 double stranded cDNA 형태로 합성 후 tagmentation 방법으로 라이브러리 제작을 수행한다(Figure 5). 최근에는 사람의 ribosomal RNA를 제거하고 순 수한 바이러스의 RNA만 분리하여 라이브러리를 제작 해
A Nextera XT transposome with adapters combined with template DNA
B Tagmentation to fragment and add adapters
www.cdc.go.kr
1182 정확도를 높이는 방법이 개발되어 본 원의 바이러스 전장 유전체
시퀀싱(whole genome sequencing)에 도입 중에 있다[5, 6].
맺는 말
국립보건연구원 염기서열 생산실에서는 최첨단 염기서열 생산 장비의 지속적 도입과 함께 최신의 라이브러리 제작 방법을 도입, 적용하여, 국내 유입 및 발생되는 병원체의 신속하고 정확한 염기서열 생산을 지원하고 있다. 향후에는 감염병 연구뿐만 아니라 병원체 염기서열 분석을 위한 라이브러리 제작 표준작업 절차서(Standard Operating Procedures; SOP)를 작성하여 제공해 나갈 예정이다.
참고문헌
1. Wotson, et al. 2013. Molecular Biology of the Gene(7th edition). Cold Spring Harbor Laboratory Press.
2. Jay Shendure, et al. Next-generation DNA sequencing. Nature Biotechnology, 2008;26(10):1135-41.
3. Steven R. Head, et al. Library construction for next-generation sequencing: O ver views and challenges. Biotechniques,
2014;56(2):61-77.
4. Ken Doyle, 2015. DNA quantitation in next generation sequencing library workflows, http://www.promega.kr/resources/pubhub/dna-quantitation-in-next-generation-sequencing-library-workflows/. 5. Christian B. M., et al. Enhanced methods for unbiased deep
sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biology, 2014;15(519):1-12.
6. Carmen F. M., et al. Efficient and unbiased metagenomic recovery of RNA virus genomes from human plasma samples. Nature,
2017;7(4173):1-13.
C. Botulinum g-DNA(before shearing)
C. Botulinum g-DNA(after shearing)
Figure 4. Size distribution of tagmentation versus physical shearing & tagmentation
Concatenated amplicon_Influenza virus
Double stranded cDNA_MERS
Figure 5. Size distribution of library according to genetic material type