저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
LDHB deficiency ameliorates ischemic
neuronal cell death
by
Song Mi Han
Major in Molecular Medicine
Department of Biomedical Sciences
The Graduate School, Ajou University
LDHB deficiency ameliorates ischemic
neuronal cell death
by
Song Mi Han
A Dissertation Submitted to The Graduate School of Ajou University in Partial Fulfillment of the
Requirements for the Degree of
Master of Biomedical
Sciences
Supervised by
Chan Bae Park, Ph.D.
Major in Molecular Medicine
Department of Biomedical Sciences
The Graduate School, Ajou University
i
- Abstract –
LDHB deficiency ameliorates ischemic neuronal cell death
Although brain occupies only 2% of body weight, it consumes approximately 25% of the body's total energy and neuronal activity comprise 80% of brain energy requirement. Thus, neurons are particularly vulnerable to situations where energy source is lacking, such as ischemia. It is well known that lactate constitutes important energy source of brain energy metabolism. How lactate supports the survival of neuronal cells is well explained by astrocyte-to-neuron lactate-shuttle hypothesis. This study focuses the role of lactate dehydrogenase B (LDHB) in neuronal cell homeostasis, which is the major enzyme in lactate-shuttle theory. For this purpose, LDHB knockout mice were generated. Both in vivo and in vitro assays show that ischemic neuronal cell death is attenuated by LDHB loss. In vivo ischemic condition, PcomA (posterior communicating artery) size of LDHB KO mice is bigger than that of WT mice. These results suggest that increased PcomA size in LDHB KO mice may be contribute to neuronal cell survival. The increase of PcomA size in LDHB KO brain is further supported by the fact that mRNA level of VEGFA (vascular endothelial growth factor A) is highly increased in LDHB KO neuronal primary cells after treatment with cobalt chloride (CoCl2), which is
hypoxia mimetic agent. Taken together, loss of LDHB might cause metabolic changes and that lead to decrease the ischemic neuronal cell death by increasing PcomA size and upregulating VEGFA level.
ii
TABLE OF CONTENTS
ABSTRACT --- i TABLE OF CONTENTS --- ⅱ LIST OF FIGURES --- iv LIST OF TABLES --- vi I. INTRODUCTION --- 1II. MATERIALS AND METHODS --- 7
A. Generation of LDHB knock-out mouse --- 7
B. Induction of global forebrain ischemia --- 7
C. Evaluation of the PcomA plasticity and tissue preparation & cresyl violet staining --- 8
D. Analysis of neuronal damage --- 8
E. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay --- 9
F. Neuron cell culture --- 9
G. Isoenzyme polymorphisms --- 10
H. MTT assay --- 10
I. Western blotting assay --- 11
J. Quantitative (q)RT-PCR--- 11
iii
III. RESULTS --- 13
1. The generation of LDHB KO mouse --- 13
2. Ischemic cell death was attenuated in LDHB KO mice via increased PcomA size in vivo --- 15
3. Hypoxia response was more upregulated in LDHB KO neuron cells after treatment with CoCl2 than LDHB expressing neuron cells --- 21
IV. DISCUSSION --- 27
V. CONCLUSION --- 30
VI. REFERENCES --- 31
iv
LIST OF FIGURES
Figure 1. Schematic representation of glucose metabolism --- 2
Figure 2. lactate astrocyte shuttle --- 4
Figure 3. Lactate dehydrogenase --- 6
Figure 4. The generation of whole body LDHB knockout mouse --- 14
Figure 5. Increased neuronal cell survival of LDHB KO mice under ischemic condition --- 17
Figure 6. Decreased TUNEL-positive fluorescence in LDHB KO neuronal cells, compared with those of WT --- 19
Figure 7. The difference of PcomA/BA (%) between LDHB WT and KO under ischemic stress --- 20
Figure 8. Culture of primary neuron cells --- 24
Figure 9. No difference of cell viability between LDHB WT and KO in vitro under hypoxia mimic condition & the conformation of hypoxia condition induced by CoCl2 --- 25
v
Figure 10. mRNA levels associated with angiogenesis after treatment with CoCl2
vi
LIST OF TABLES
Table 1. Primer information of PCR ---12
I. Introduction
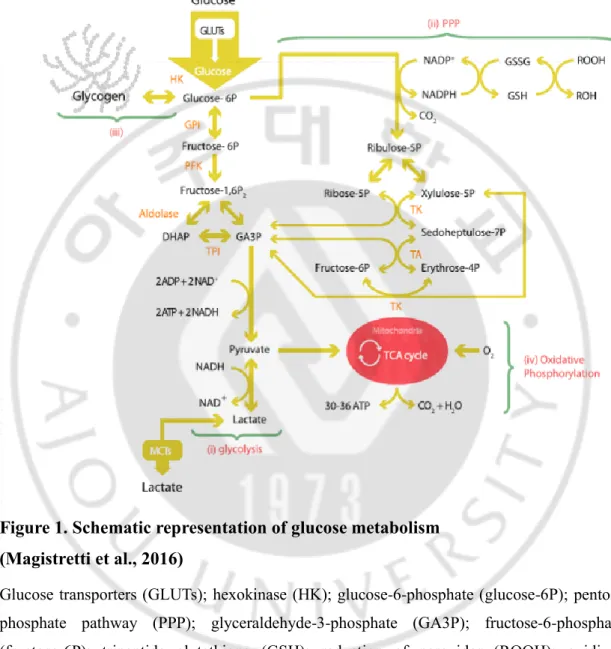
Brain is the most energy consuming organ in human body. Brain weighs 2% of the body weight, but consumes about 25% of total energy. Glucose is the main source to meet brain’s energy demands (Newington et al., 2013). In the brain, 6.5 mg glucose per 100 g human brain tissue is consumed per minute (Mergenthaler et al., 2013). Glucose is transported by glucose transporter (GLUTs) from blood to brain, and then it is transformed to glucose-6-phosphate by hexokinase. Glucose-6-phosphate is metabolized through 1) glycolysis 2) pentose phosphate pathway (PPP) 3) glycogenesis and 4) oxidative phosphorylation (Figure 1) (Bélanger et al, 2011). Brain is composed of many cells, including neurons, glial and ependymal cells. Each brain cell types have different function and metabolic requirements and the expression patterns of their key genes regulating energy metabolism are distinct (Felipe et al., 2012; Newington et al., 2013).
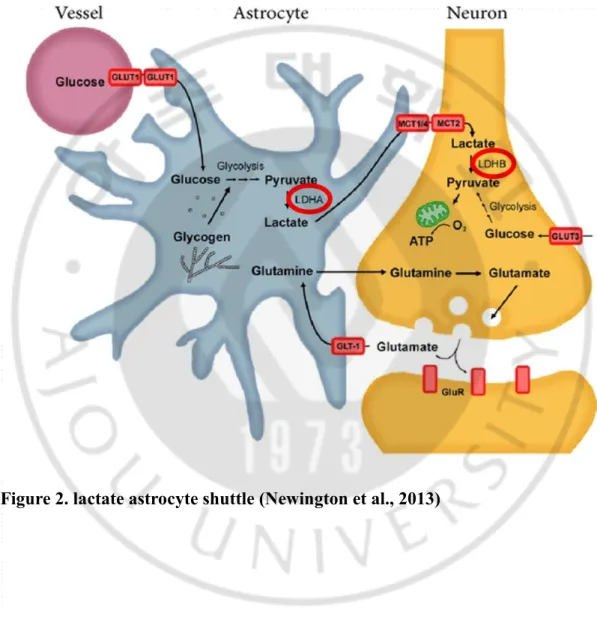
Among different brain cells neurons consume the most energy which comprises about 80% of brain energy expenditure, while other cell types consume the rest (Bélanger et al, 2011; Mergenthaler et al., 2013; Felipe et al., 2012). Interestingly, astrocytes take up 80% glucose from the brain parenchyma, while neurons take up only 20% glucose (Magistretti et al., 2011). Because of this bias in glucose consumption, it suggests that astrocyte and neuron interact with each other. This perspective is reasonably explained by astrocyte-to-neuron lactate-shuttle hypothesis (ANLSH) (Rho, 2015). ANLSH insists that astrocyte metabolize glucose to lactate, and then lactate is transferred to neurons as energy substrate (Rho, 2015). It is well known that astrocytes have high glycolytic activity, so they metabolize glucose and produce lactate very quickly (Falkowska et al, 2015). On the other hand, neurons tend to prefer lactate to glucose as energy source, even though both lactate and glucose are
present (Pellerin et al, 2012).
Figure 1. Schematic representation of glucose metabolism (Magistretti et al., 2016)
Glucose transporters (GLUTs); hexokinase (HK); glucose-6-phosphate (glucose-6P); pentose phosphate pathway (PPP); glyceraldehyde-3-phosphate (GA3P); fructose-6-phosphate (fructose-6P); tripeptide glutathione (GSH); reduction of peroxides (ROOH); oxidized glutathione GSSG); glucose-6-phosphate isomerase GPI); phosphofructokinase (PFK); fructose-1,6-bisphosphate fructose-1,6-P2); dihydroxyacetone phosphate (DHAP); triose phosphate isomerase (TPI); ribose-5-phosphate (ribose-5P); xylulose-5-phosphate (xylulose-5P); transketolase (TK,); sedoheptulose-7-phosphate (sedoheptulose-7P); transaldolase (TA); erythrose-4-phosphate (erythrose-4P).
A main concept in brain metabolism is the astrocyte-to-neuron lactate-shuttle hypothesis (ANLSH) (Rho, 2015). The astrocyte-derived lactate is the main substrate that meets the energetic expenditure of neurons (Pellerin et al., 2012). Glucose transporters (GLUTs), lactate transporters (monocarboxylate transporters, MCTs), and lactate dehydrogenases (LDHs) are expressed by astrocytes and neurons, but astrocytes and neurons express different isozyme of each transporters (Genc et al., 2011). Astrocytes take up glucose through GLUT1, while glucose in neuron is transmitted via GLUT3 (Newington et al., 2013). GLUT1 and GLUT3 have different kinetic properties: GLUT3 is a high affinity and high capacity glucose transporter compared to GLUT1 (Shah et al., 2012). Therefore, the uptake of glucose or lactate by neurons depends on the location and needs of the cell (Newington et al., 2013). Glucose taken up via this mechanism is used to produce lactate glycolytically. That lactate released from astrocyte by MCT4 and MCT1 is taken up into neuron by MCT2 (Magistretti et al., 2015). MCT plays a role as proton-linked membrane carrier that transports monocarboxylates including lactate, pyruvate and ketone bodies across the membrane (Suzuki et al., 2011). The astrocyte-derived lactate is oxidized to pyruvate, which is then metabolized in mitochondria to produce ATP (Magistretti et al., 2015).
The key molecule of astrocyte-to-neuron lactate-shuttle theory is lactate dehydrogenase (LDH). (Rho, 2015). Lactate dehydrogenase (LDH) is an intracellular enzyme found in various human tissues such as brain, skeletal muscle, heart, liver and kidneys, and mediates the inter-conversion of pyruvate and lactate (P R Sharma et al, 2010). LDH is a tetrameric enzyme composed of two subunits, LDHA (subunit M) and LDHB (subunit H), which is combined to form five LDH isozymes (Russell, 2002). Five LDH isozymes (H4 or LDH-1, H3M or LDH-2, H2M2 or LDH-3, HM3 or LDH-4, and M4 or LDH-5) are determined by the ratio of LDH-5 to LDH-1 subunits. (Philippe, 1996). The LDH-5 subunit prefers to form lactate from pyruvate, while the LDH-1 subunit has a preference of the reaction which is the formation of pyruvate from lactate. LDH isozymes express in many kinds of organs and tissues (M. Drent et al, 1996). In brain, astrocytes mainly express LDHA, while neurons predominate LDHB (Albert et al. 2015).
According to ANLSH, lactate is regarded as the main energy source of neuronal cells. Especially, lactate is acknowledged not only as an important energy substrate for neurons but as a preferred source of energy under certain circumstances like hypoxic condition (Newington et al., 2013). Here, I investigate whether LDHB contributes to neuronal energy metabolism and neuronal cell survival. I show that LDHB deficiency leads to resistance of neuronal cells to ischemic condition. Unexpectedly, there was no difference between absence and presence of LDHB in normal condition. Surprisingly, LDHB KO mice show expanded in size of the posterior communicating artery (PcomA), comparing to that of normal mice. Increased PcomA size might positively affect to survival of neuron cells in ischemic condition. It is also supported by in vitro assay using a primary neuron cells with or without LDHB expression. In vitro assay also shows that hypoxia response, including vascular endothelial growth factor A (VEGFA), is more upregulated in LDHB non-expressing neuronal cells, compared with LDHB expressing neuronal cells.
II. MATERIALS AND METHODS
A. Generation of LDHB knock-out mouse
A targeting vector contains loxP sites flanked LDHB and Frt-PGK-NEO-Frt cassette (Frt-site-flanked neomycin gene expressed from the phosphoglycerate kinase promoter). The targeting vector was transfected to the embryonic stem cells (ES cells). The targeting vector replaced the ES cells endogenous DNA by homologous recombination. The Frt-PGK-neo-Frt gene was excised by mating LDHB-neo- loxP mice with transgenic Flp-recombinase expressing mice. The LDHB knockout mice were generated by crossing 2 loxP sites flanked LDHB mice with protamine-cre recombinase carrying mice.
PCR analysis was used to identify genotype of WLDHB mouse (WLDHB knock-out primer: the forward primer, 5′- TCATGGCAGGGACAGCACAGT-3′; the reverse primer, 5′-TCAAGTCCCAGGATAGTCATAGC-3′ / LDHB primer: the forward
primer, 5′-CATTGGGTGCTTAGCCATTT-3′; the reverse primer,
5′-AAACACAAAACCAGCCGTTC-3′).
To generate the embryo (LDHB WT; KO), LDHB WT homozygote or LDHB KO homozygote were mated overnight and checked for the plug the following morning, respectively. We sacrificed embryos 14 days after plug checked.
B. Induction of global forebrain ischemia
Mice were anesthetized 1% halothane in a 70% nitrous oxide and 30% oxygen mixture using a face mask during bilateral common carotid artery occlusion (BCCAO) for 10min. Through midline incision, both common carotid arteries were exposed and were temporarily clamped with micro serrefines. After reperfusion, the animals were cared in a warm incubator (32~33℃) until they were sacrificed three days later. During the surgical procedure, rectal temperature was maintained between
36.5~37.9℃ with a heating lamp.
C. Evaluation of the PcomA plasticity and tissue preparation & cresyl violet staining
Three days after transient forebrain ischemia, the animals were transcardially perfused with 4% paraformaldehyde in 1XPBS, followed by 1 mL of latex solution to evaluate the plasticity of the posterior communicating artery. After the mice had been cooled on ice for 2 hour, the brains were isolated and photographed with a digital camera (EOS 300D, Canon, Tokyo,Japan). They were then post-fixed in 4% paraformaldehyde for 24 h and cut using a cryotome (20 μm sections) after they had been dehydrated with 30% sucrose in 0.1 M PB.
For cresyl violet staining, the sections were mounted on coated slides and allowed to air-dry overnight. The mounted sections were rehydrated in distilled water, and incubated with 0.1% cresyl violet solution for 10 min. The sections were rinsed in 70% ethanol and dehydrated in graded series of ethanol, immersed in xylene, mounted with permount and cover slipped.
D. Analysis of neuronal damage
The severity of ischemic neuronal damage in the medial CA1 area of the hippocampus (Figure 8) was evaluated by an examiner blinded to the study groups and semi quantitatively as sessed according to the method described previously (Murakami et al, 1998; Choet al, 2006). Ischemic neuronal damage was measured on the following scale: no ischemically damaged neurons (grade 0), less than 24% ischemic neuronal damage (grade 1), 26∼50% ischemic neuronal damage (grade 2), 51∼75% ischemic neuronal damage (grade 3), 76~100% of ischemic neuronal damage (grade 4).
E. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay
Cell death was detected by TUNEL assay using DeadEndTM Fluorometric TUNEL
System (Promega, G3250). The brain tissue section was prepared in slides. The tissue slides were fixed with 4% paraformaldehyde for 10 minutes. The fixed tissue slides were equilibrated in 100 μl equilibration buffer at room temperature for 5 minutes, after immersing in ethanol:acetic acid=2:1 at – 20 ℃ for 5 minutes. And then the slides were incubated with 51 μl (equilibration buffer 45 μl + nucleotide mix 5 μl + terminal deoxynucleotide transferase recombinant (rTdT) 1 μl) per one slide at 37 ℃ for 1 hour. For stopping the reaction, the slides were immersed in 2XSSC for 15 minutes. Finally, the slides were washed in PBS, 5 minutes each time and added mounting medium. Then, the slides were incubated with DAPI solution to visualize all nuclei. The death of neuron cells in brain were photographed under a fluorescence microscope.
F. Neuron cell culture
For primary neuron cultures, cortex and hippocampus were isolated from LDHB WT and knockout mouse [LDHB (+/+); LDHB (-/-)], E14 embryos. Cortex and hippocampus were separated from mouse, and then centrifuged at 1,300 x rpm for 3 minutes. Then supernatant was removed, and pellet was mildly resuspended several times in 1 mL plating medium (PM) using a flame-polished Pasteur pipette. To seed with 1 X 106 cell/mL, cells were counted after stained with trypan blue and then cells
were cultured in poly-D-lysine solution-coated (PDL coated) plates. MEM (minimum essential media) supplemented with 200 mM glucose) was used for isolation of the cortex and hippocampus. PM (MEM added with 200 mM glutamine, 5% FBS (Fetal bovine serum), 5% HS (horse serum)) was utilized for plate of these cells. Cultured neuron cells were added with 5 μM ARA-C dissolved in growth media (GM) (MEM added with 200 mM glutamine, 5% HS (horse serum)) at 2 days, and further treated
with CoCl2 at 5 days after plated.
For confirming neuron cells, immunocytochemistry (ICC) is performed. Neuron is stained by anti-NeuN which is a neuron marker. And ICC sample is identified by fluorescence microscopy.
G. Isoenzyme polymorphisms
Isoenzyme patterns of lactic dehydrogenase (LDH) were detected using vertical slab discontinuous native polyacrylamide gel electrophoresis (Native-PAGE) assay. The neuron cells were harvested, and protein extraction solution (20 mM Tris (pH 7.4), 5 mM EDTA, 120 mM KCl, protease inhibitor cocktail) was added. Then the mixture was sonicated by bioruptor and centrifuged at 13,000 x rpm for 10 minutes. The supernatant was aliquoted and stored at -80°C. Liquid sucrose [40% (m/v)] and the samples were mixed [1:1 (v/v)] and then loaded into the individual lanes of the polyacrylamide gel. Isoenzyme patterns were detected by activity staining using NBT/PMS under dark and the gels were fixed with 5% acetic acid. The LDH isoform bands were scanned.
H. MTT assay
The cells were plated into 48-well plates at a density of 2 x 105 cells/well. Following
overnight incubation, the cells were treated with CoCl2 for 12 h. A total of 20 µL MTT
solution (5 mg/ml) was subsequently added to each well and incubated for 4 h at 37˚C. 200 µL MTT solvent (4 mM HCl, 0.1% NP-40 in isopropanol) were placed in each well after removal of medium. Following agitation for 10 min, the absorption value of each well was measured at 595 nm using a microplate reader. Absorbance at 595 nm was normalized to that of untreated control (100%), and the results were expressed as viability of control. All experiments were performed in triplicate.
I. Western blotting assay
Neuron cells were grown in PDL-coated 6-well or 12-well plates and cultured for 5 days. NPCs were lysed in lysis buffer (RIPA buffer (BIO BASIC CANADA INC)), containing proteinase inhibitor cocktail. The cell lysates were further homogenized by brief sonication and centrifuged at 13,000 rpm for 10 minutes at 4°C. The resulting supernatant was collected.
Bradford protein assay (DC™ Protein Assay, BIO-RAD) was used to quantify the total protein content. Samples containing equal quantities of protein were resolved using 10-12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes. The membrane was blocked with 5% BSA and incubated with primary antibodies targeting HIF-1α (Cayman (10006421) 1:1,000), LDHA (Abcam (ab9002) 1:2,000), LDHB (Abcam (ab85319) 1:2,000) and AGE (cell biolabs (STA-011) 1:1,000) for overnight. After washing with PBS containing Tween-20 (PBST), the membranes were subsequently incubated with peroxidase-conjugated secondary antibodies for 1 h. Protein bands incubated with enhanced chemiluminescence reagents (advansta) were visualized using Developer. To ensure equal protein loading, each membrane was reprobed with anti-tubulin antibody (1:5000). Tubulin was used as a control for protein level quantification.
J. Quantitative (q)RT-PCR
Total RNA was extracted from cultured primary neuron cells using RNeasy extraction kit (Qiagen) according to the protocol supplied with the kit. For cDNA synthesis, High capacity cDNA archive kit (ABI) was used.
qRT-PCR analysis was carried out using Sso Advanced SYBR Green Supermix (Bio-Rad). The double △Ct method was employed to calculated the relative expression of target gene: Relative expression of target gene = 2−△△Ct, △Ct = Ct (Target Gene) −Ct (Control); △(△Ct) = △Ct (sample)−△Ct (control).
To inquiry the mRNA sequences of target genes (HIF1-α; VEGFA; VEGFB; VEGFC; Angpt1; Angpt2; brain angiogenesis inhibitor-1(BAI1)), we designed the real-time PCR primers using NCBI database. All primers were synthesized by M-Biotech Corporation. The specific sequences were shown in the Table 1. PCR reaction system was shown in Table 2.
Table 1
Primer information of PCR
Target genes Primer Primer’s Sequence (5’-3’)
VEGFA forward primer CGTCAGAGAGCAACATCACCA
reverse primer GCGCTTTCGTTTTTGACCCT
ADGRB (BAI1)
forward primer CCCAGCTCATGACCGACTTT
reverse primer TTAGCTGGCAGGCTGTTGAA
Table 2
PCR reaction system and amplification temperature cycle parameters PCR reaction system and amplification temperature cycle parameters
Components Volume per reaction
Sso Advanced SYBR Green Super mix 10 μL
Forward primer (10 pM) 1 μL
Reverse primer (10 pM) 1 μL
cDNA template 8 μL
K. Data analysis
Results were reported as means ± SEM. Data were analyzed by using a one-way ANOVA and Bonferroni t test. Statistical significance was determined if P ≤ 0.05.
III. RESULTS
1. The generation of LDHB KO mouse
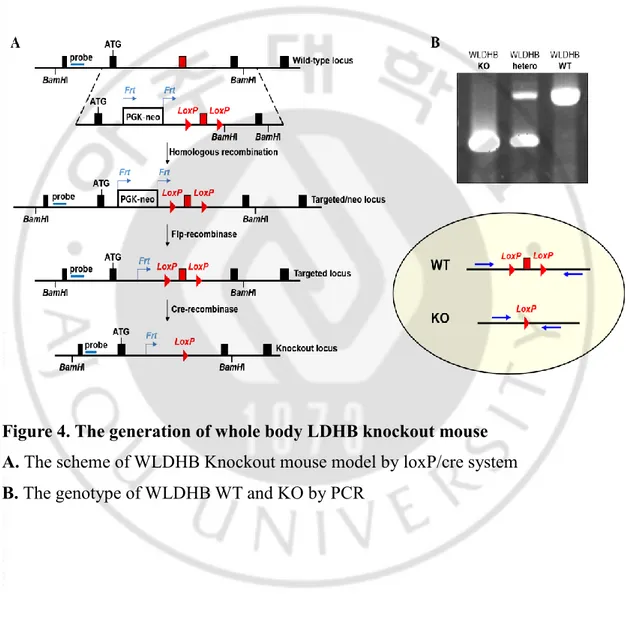
Whole body LDHB gene knockout mouse is generated by inactivation of LDHB gene by cre/loxP system (Figure 4A). The LDHB gene which has flanked by two loxP sites can be excised and inactivated by Cre recombinase recognizing two loxP sites.
Two mouse lines were required for conditional gene-LDHB deletion. First, a conventional transgenic mouse line with Cre targeted to protamine. Secondly a mouse strain that has a target gene–LDHB flanked by two loxP sites. Recombination occurs only in those cells expressing Cre recombinase. Thus, the LDHB remains active in all cells and tissues which do not express Cre recombinase. We successfully generated whole body LDHB knockout mouse. The whole body LDHB knockout mouse (WLDHB KO) showed 403 bp size band, while the whole body LDHB wild type mouse (WLDHB WT) showed 660 bp size band by PCR (Figure 4B).
Figure 4. The generation of whole body LDHB knockout mouse A. The scheme of WLDHB Knockout mouse model by loxP/cre system B. The genotype of WLDHB WT and KO by PCR
2. Ischemic cell death was attenuated in LDHB KO mice by PcomA size in vivo
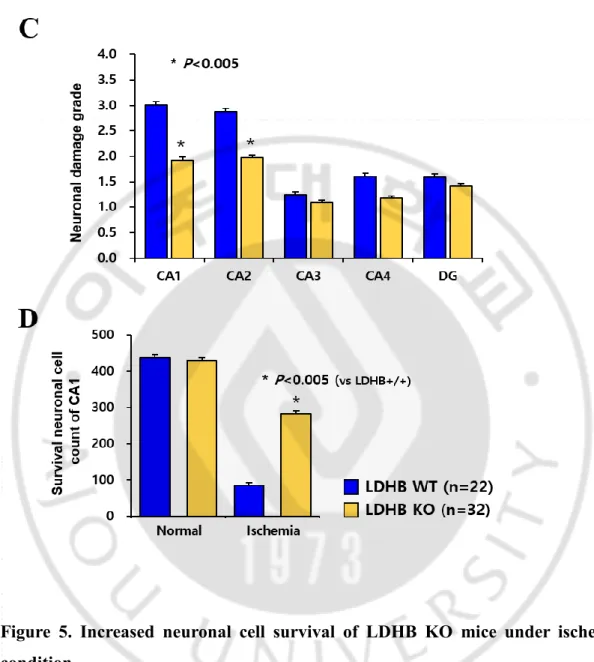
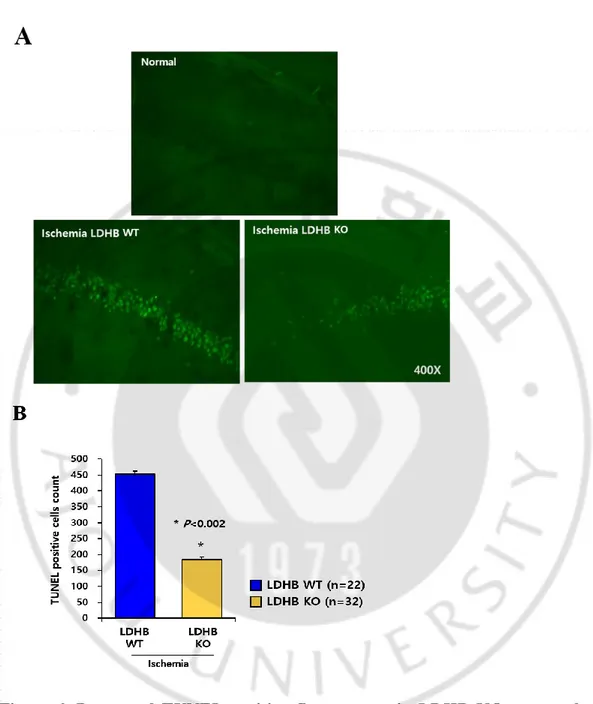
In brain, cells consume large amounts of metabolite and energy substrate, including glucose, lactate, and ketone. Especially lactate is the main energy source in neuron cells and it should transfer to pyruvate before oxidized for ATP production. For this reason, LDHB may play a crucial role on neuronal metabolism. To investigate whether LDHB contributes to neuron cell survival, histological staining using cresyl violet was performed. Interestingly, there was no phenotypical change by LDHB KO, including neuronal cell number, in normal condition (Figure 5A, upper panel). It is assumed that LDHB KO neurons may metabolically adapted. So, I further examined in metabolic stress condition like ischemic condition for observing dramatical change by LDHB. For this purpose, bilateral common carotid occlusion (BCCO) in the mouse is utilized as a common ischemic model, in terms of mortality and susceptible neuronal degeneration of CA1 region (Barone,1993). As shown in Figure 5, LDHB KO mice was resistant to neuronal cell death in ischemic condition. The results of histological staining using cresyl violet showed that ischemic stress effectively induced cell death in neuron cells of LDHB WT mice, but not those of KO mice (Figure 5A). In addition, ischemic damage in neuron cells was evaluated using the following scale: no damaged, and every quarters are expressed to grade 0 (0%), grade 1 (up to 24%), grade 2 (25% - 50%), grade 3 (51% - 75%), and grade 4 (upper than 75%), respectively (Figure 5B). Furthermore, when survival neuron cells of CA1 region were counted and described to graph, these results showed that neuronal cell survival was markedly increased in LDHB deficient mice (Figure 5C). To confirm preceding data, TUNEL assay was performed in ischemic condition. Ischemic condition outstandingly induced TUNEL fluorescence in neuronal cells of LDHB WT mice, whereas TUNEL-positive cells were decreased in those of LDHB KO mice (Figure 6A, B). I drew TUNEL-positive cells graphically by counting cells (Figure 6B). Collectively, I found that deficiency of LDHB contributes to cell survival ability
of neuronal cells and resistance to ischemic cell death.
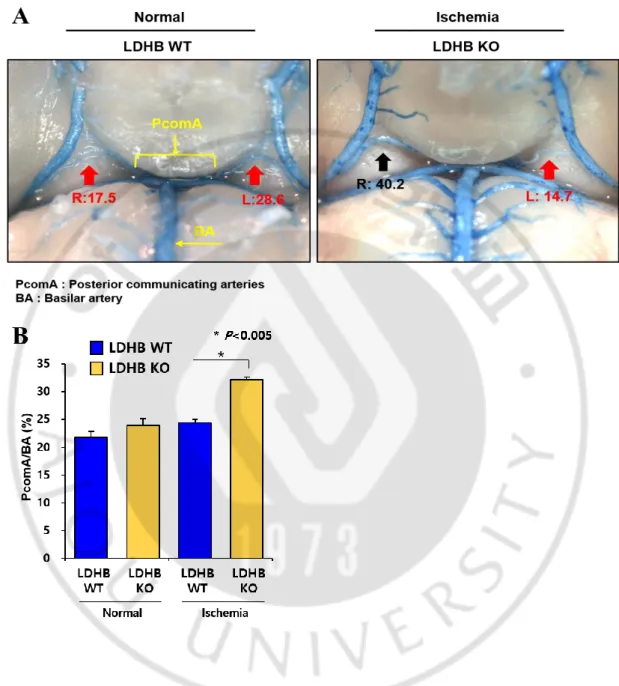
The patency of the PcomA is regarded as a crucial factor in ischemia after bilateral common carotid artery occlusion (BCCO) (Kitagawa et al., 1998). Commonly, neuron cell death is induced at less than 30% PcomA/BA (basilar artery) (Cho, 2006). While there is no difference of the PcomA size between WLDHB WT and WLDHB KO in normal condition, it markedly distinguished in ischemic condition (Figure 7). LDHB KO mice having over 30% PcomA/BA was more than WLDHB KO mice after tBCCAO. These in vivo results suggest that the increase of PcomA size in WLDHB KO mice affects neuron cell survival in ischemic condition.
Figure 5. Increased neuronal cell survival of LDHB KO mice under ischemic condition
A. Cresyl violet staining of the fixed hippocampus isolated from the mouse after ischemic stress
B. Neuronal damage grades of the hippocampus C. Survival neuron cell count in CA1 region
Figure 6. Decreased TUNEL-positive fluorescence in LDHB KO neuronal cells, compared with those of WT
A. TUNEL staining of the fixed hippocampus isolated from the mouse after ischemic stress
Figure 7. The difference of PcomA/BA (%) between LDHB WT and KO under ischemic stress
A. Representative images of latex dye casting show PcomA B. The graphical description of PcomA/BA (%)
3. Hypoxia response was more upregulated in LDHB KO neuron cells after treatment with CoCl2 than LDHB expressing neuron cells
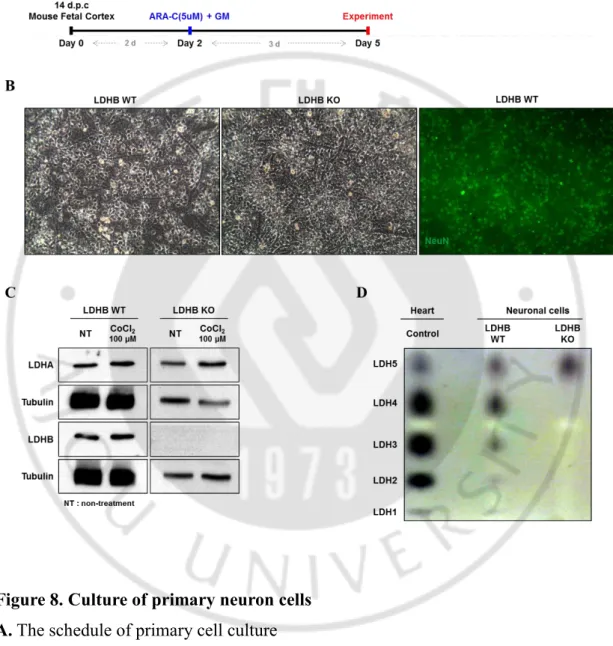
For neuron primary culture in vitro, first I mated mice WLDHB WT female/male and KO female/male, respectively. Fourteen days after plug checked, I cultured primary neuron cells using cortex and hippocampus isolated from embryos. Additionally, 2 days after culture, cytosine arabinoside (AraC) was treated for 3 days to decrease glial cell contamination, which was known as anti-mitotic agent reducing the population of non-neuronal cells capable of DNA synthesis (Seibenhener et al. 2012). After treatment, the neuronal cells were used to perform following experiments (Figure 8 A). For confirming neuron cells, immunocytochemistry was performed using specific antibody targeting NeuN proteins, neuron marker protein (Figure 8 B). Similar to in vivo data, in vitro cells, phase-contrast microscopy showed that there is no difference between primary neuron cells from WLDHB WT mice and those of WLDHB KO mice (Figure 8 B).
To confirm genetic type of primary neuron cells, protein was extracted from each cultured primary neuron cells and then western blot was performed (Figure 8 C). The reason of increasing LDHA in hypoxia condition is that LDHA is the downstream target of HIF-1α. Furthermore, since LDH is homo- or hetero-tetrameric proteins, isoenzyme pattern analysis was performed. To confirm isoenzyme patterns, protein lysates were isolated from both WT and KO neuron cells using specific protein extraction solution which is not contain detergent and reducing agent and separated by Native-PAGE. LDH isozyme is separated by their charge. LDHB carry more negative charges than LDHA. So, LDHA is appeared more upper than LDHB. When isoenzyme patterns were detected by activity staining using NBT/PMS under dark, only LDH5 was expressed in KO neuron cells, which is LDHA only homo-tetrameric enzyme (Figure 8 D).
Hypoxia inducible factor-1 (HIF-1) is composed of HIF-1α and HIF-1β subunit as a heterodimeric transcription factor (Zhou J et al, 2004). HIF-1β is constitutively present and its mRNA and protein level is sustained constantly regardless of oxygen availability, while HIF-1α has a short half-life and its stability is controlled by oxygen (Ke et al. 2006). In normoxia, HIF-1α exists low level du to continuous degradation by the 26S proteasome, whereas under hypoxia, HIF-1α becomes stabilized by blocking prolyl hydroxylases (HIF-PH). In this way, many chemicals as transition metals such as CoCl2, or the iron chelator desferroxamine (DFX) are widely used by
blocking HIF-PH and in turn inducing HIF-1α stabilization (Zhou J et al, 2004). So, to mimic ischemic condition in vitro, I treated cobalt chloride (CoCl2), which
enhance the stability of hypoxia-inducible factor (HIF)-1α, in the neuron cell (Zhang et al. 2014). After treating CoCl2 for 12 hour, MTT assay is used for identification of
neuron cell viability (Figure 9 A). The decreasing viability of LDHB KO neuron cells after treatment with CoCl2 was similar to that of LDHB WT neuron cells. These
results suggest that resistance of LDHB KO mice to neuronal cell death under ischemic condition might be irrelevant to cellular level. Rather, PcomA size might play crucial rules on resistance to ischemic stress. To identify the reason of resistance to ischemic stress, I further tested whether proteins associated with hypoxia response were activated by CoCl2 in LDHB KO neuron cells. Even though HIF-1α protein level
was dramatically induced by CoCl2 in protein extracts of neuron cells isolated from
LDHB WT and KO (Figure 9 B), its target gene, VEGFA, was significantly increased in neuronal cells of LDHB KO, more dramatically than those of WT (Figure 10). On the other hand, brain-specific angiogenesis inhibitor 1 (BAI 1) displayed a tendency to decrease in LDHB KO cells compared to LDHB WT cells (Figure 10). Increase of the VEGF expression by HIF-1 and decrease of the BAI 1 could induce the development of new blood vessels of the target area in the brain. The production of new blood vessels leads to provide an increased blood flow and oxygen supply and that results in reducing ischemic damage (KE et al., 2006; Tulsulkar et al., 2013).
Taken together, these results suggest that resistance of LDHB KO to neuronal cell death under ischemic condition is derived from upregulated response to hypoxia prior to PcomA size increase.
Figure 8. Culture of primary neuron cells A. The schedule of primary cell culture
B. Morphology of neuronal cells and the confirmation of neuron cell by ICC C. The western blot of the extracted protein comparing LDHB KO with WT D. LDH isoenzyme pattern comparing LDHB KO with WT
Figure 9. No difference of cell viability in vitro under hypoxia mimic condition & the conformation of hypoxia condition induced by CoCl2
A. Cellular viability using MTT assay after treatment of primary neuronal cells with CoCl2
B. Western blotting of HIF1a, LDHA and LDHB was performed in LDHB WT, KO neuron cells. Tubulin was used as a loading control in western blots
Figure 10. mRNA levels associated with angiogenesis after treatment with CoCl2
VEGF-A, BAI 1 mRNA levels through Real-Time PCR after extracting cDNA of WT, KO neuron cells.
IV. DISCUSSION
In this study, I found that LDHB deficiency increase neuronal cell survival under ischemic condition in vivo. LDHB null mice did not show any different phenotype compared with wild type mice (Figure 5 A). In parallel, no morphological difference also was observed in primary neuronal cells isolated from cortex and hippocampus of mice having LDHB mice (Figure 8 B). But in metabolic stress condition, absence of LDHB increased the cell survival ability via increase of PcomA size. When I performed the histological staining using a cresyl violet to detect survival cells in ischemic condition, decrease of live cells was observed in only WT mice, but not KO mice (Figure 5 A). In addition, when I further examined TUNEL-assay to detect dead cells, TUNEL-fluorescence positive cells were outstandingly induced in only WT mice (Figure 6). These results indicated that loss of LDHB positively affects cell viability and increase of PcomA size (Figure 7) might be reasonable explain. When I treated of primary neuronal cells with CoCl2 to mimic ischemic condition in cell
culture system, the cell viability is no different between LDHB WT and KO (Figure 9). But, in ischemic condition, VEGFA was effectively increased in LDHB KO neuron cell (Figure 10). Taken together, in ischemic stress condition, deficiency of LDHB protects ischemic neuronal cell death by increasing PcomA size and VEGFA level. The relationship of VEGFA and PcomA is unclear. But, VEGFA may be also relevant to vasodilation, although VEGFA is well known as the role of mediating angiogenesis and vasculogenesis. I will further investigate the mechanism of vessel dilation in LDHB KO brain. In ischemic condition, HIF-1α expression is increased and in turn, increased HIF-1α induce production of VEGFA. In addition, VEGFA stimulates endothelial nitric oxide synthase (eNOS) which produces nitric oxide (NO). NO is a crucial mediator of blood vessel reactivity, angiogenesis and vasodilation (Johanna et al., 2012). So, it is expected that VEGFA may be involve in increasing PcomA size.
And then, further study is needed to confirm why PcomA size is increased in LDHB deficiency under ischemic condition and how PcomA size is regulated.
Metabolic adaptation also is in close connection with ischemic cell death (Michelle et al., 2009). LDHB is most important enzyme for energy metabolism of neuronal cells, according to astrocyte-to-neuron lactate-shuttle theory. Therefore, LDHB deficiency may induce metabolic adaptation, such as increased capillary surface area that results in increased oxygen delivery, decreased energy demand and increased energy production efficiency, and this metabolic adaptation might contribute to resistance of neuronal cells to cell death under ischemic stress. LDHB deficiency mice also may be involved in the ischemic precondition which is resistance to the loss of blood supply and oxygen. To confirm whether ischemic precondition is induced by LDHB deficiency, further studies are needed.
In the ischemic condition, mitochondrial dysfunction is considered as key cause of neuronal cell death (Baxter et al., 2014; Niizuma et al., 2009). Therefore, mitochondrial oxygen consumption rates, ROS levels, ATP levels, and mitochondrial membrane potential should be examined using mice and its primary cultured neuron cells, having or not LDHB gene, under ischemic stress.
In brain, increasing activity is accompanied by changes in local blood flow and glucose utilization being referred to as neurovascular and neurometabolic coupling (Bélanger et al., 2011). So, brain energy metabolism has relevance to vascular and metabolic sources.
Here, I found that ischemic neurons of LDHB deficiency mice survive well by the increased PcomA size. Although more studies are need for the mechanism of increasing PcomA size, LDHB may be related to vasodilation. So, I suggest that LDHB has the potential of remedial agent as vasodilator for treating the diseasesthe blood vessels get blocked such as stroke and cerebral infarction. And I also expect that LDHB is helpful for surviving neurons under ischemic condition. So, LDHB may be possible to be a good candidate for treating neurodegenerative disease such as
epilepsy, stroke, Alzheimer’s disease, aging, ischemia and dementia (Sada et al., 2015; Rho, 2015).
V. CONCLUSION
In this study, I demonstrated that LDHB KO mouse ameliorate ischemic neuronal cell death through the increase of PcomA size which was supported by the fact that VEGFA level is upregulated I cells. LDHB is an important enzyme in neuron for using lactate as energy source by converting lactate to pyruvate, since neuron uses lactate more than glucose as energy source. In normal condition, LDHB KO neuron cell appeared no difference, but in ischemic condition, the decreased neuronal cell death is shown. That means LDHB KO neuron cell may be less vulnerable to ischemia by preconditioning compared to LDHB WT. To elucidate preconditioning in LDHB KO neuron cells, further studies are needed.
VI. REFFERENCES
1. Albert Gjedde, Pierre Magistretti “Cellular Mechanisms of Brain Energy Metabolism” Neurosourgery. 2015
2. Barone FC, Knudsen DJ, Nelson AH, Feuerstein GZ, Willette RN. “Mouse Strain Differences in Susceptibility to Cerebral Ischemia Are Related to Cerebral Vascular Anatomy.” J Cereb Blood Flow Metab. 13(4):683-92. 1993. 3. Baxter P, Chen Y, Xu Y, Swanson RA. “Mitochondrial dysfunction induced by nuclear poly(ADP-ribose) polymerase-1: a treatable cause of cell death in stroke.” Transl Stroke Res. 5(1):136-44. 2014.
4. Bélanger M, Allaman I, Magistretti PJ “Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation” Cell Metab. 7;14(6):724-38. 2011. 5. Bittar PG, Charnay Y, Pellerin L, Bouras C, Magistretti PJ. “Selective
distribution of lactate dehydrogenase isoenzymes in neurons and astrocytes of human brain.” J Cereb Blood Flow Metab. 16(6):1079-89. 1996.
6. Crawford RM, Budas GR, Jovanović S, Ranki HJ, Wilson TJ, Davies AM, Jovanović A. “M-LDH serves as a sarcolemmal K(ATP) channel subunit essential for cell protection against ischemia.” EMBO J. 1;21(15):3936-48. 2002.
7. Drent M, Cobben NA, Henderson RF, Wouters EF, van Dieijen-Visser M. “Usefulness of lactate dehydrogenase and its isoenzymes as indicators of lung damage or inflammation.” Eur Respir J. 9(8):1736-42. 1996.
8. Falkowska A, Gutowska I, Goschorska M, Nowacki P, Chlubek D, Baranowska-Bosiacka I “Energy Metabolism of the Brain, Including the
Cooperation between Astrocytes and Neurons, Especially in the Context of Glycogen Metabolism" Int J Mol Sci. 29;16(11):25959-81. 2015.
9. Felipe A. Beltrán, Aníbal I. Acuña, María Paz Miró and Maite A. Castro
“Brain Energy Metabolism in Health and Disease”ISBN 978-953-51-0207-6.
2012.
10. Genc S, Kurnaz IA, Ozilgen M. “Astrocyte-neuron lactate shuttle may boost more ATP supply to the neuron under hypoxic conditions--in silico study supported by in vitro expression data.” BMC Syst Biol. 5:162. 2011.
11. Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. “Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain.” Stroke. 32(5):1208-15. 2001.
12. Johanna La¨hteenvuo, Anthony Rosenzweig “Effects of Aging on Angiogenesis” Circulation Research. 110:1252-1263. 2012.
13. Ke Q, Costa M. “Hypoxia-inducible factor-1 (HIF-1).” Mol Pharmacol. 70(5):1469-80. 2006.
14. Kitagawa K, Matsumoto M, Yang G, Mabuchi T, Yagita Y, Hori M, Yanagihara T. “Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: evaluation of the patency of the posterior communicating artery.” J Cereb Blood Flow Metab. 18(5):570-9. 1998.
15. Kyung-Ok Cho, Seul-Ki Kim, Young-Jin Cho, Ki-Wug Sung, and Seong Yun Kim. “A Simple Method for Predicting Hippocampal Neurodegeneration in a Mouse Model of Transient Global Forebrain Ischemia.” Korean J Physiol Pharmacol 10(4): 167-172. 2006.
Metabolism and Functional Imaging” Neuron. 20;86(4):883-901. 2015.
17. Magistretti PJ. “Neuron–glia metabolic coupling and plasticity” Exp Physiol. 96(4):407-10. 2011.
18. Mergenthaler P, Lindauer U, Dienel GA, Meisel A. “Sugar for the brain: the role of glucose in physiological and pathological brain function” Trends Neurosci. 36(10):587-97. 2013.
19. Michelle A. Puchowicz, Smruta S. Koppaka, Joseph C. LaManna. “Brain Metabolic Adaptations to Hypoxia” Metabolic Encephalopathy, 15-30, 2009 20. Newington JT, Harris RA, Cumming RC “Reevaluating Metabolism in
Alzheimer's Disease from the Perspective of the Astrocyte-Neuron Lactate Shuttle Model.” J Neurodegener Dis. 2013:234572. 2013.
21. Niizuma K, Endo H, Chan PH. “Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival.” J Neurochem. 109 Suppl 1:133-8. 2009.
22. Pellerin L, Magistretti PJ. “Sweet sixteen for ANLS.” J Cereb Blood Flow Metab. 32(7):1152-66. 2012.
23. Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. “Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review.” Front Pharmacol. 25;2:49. 2011.
24. Rho JM “Inhibition of Lactate Dehydrogenase to Treat Epilepsy” N Engl J Med. 373(2):187-9. 2015.
25. Sada N, Lee S, Katsu T, Otsuki T, Inoue T. “Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy.” Science. 20;347(6228):1362-7. 2015.
from prenatal mice.” J Vis Exp. 26;(65). pii: 3634. 2012.
27. Shah K, Desilva S, Abbruscato T. “The role of glucose transporters in brain disease: diabetes and Alzheimer’s Disease.” Int J Mol Sci. 3;13(10):12629-55. 2012.
28. Sharma PR, Jain S, Bamezai RN, Tiwari PK. “Utility of serum LDH isoforms in the assessment of mycobacterium tuberculosis induced pathology in TB patients of Sahariya tribe.” Indian J Clin Biochem. 25(1):57-63. 2010.
29. Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. “Astrocyte-neuron lactate transport is required for long-term memory formation.” Cell. 4;144(5):810-23. 2011.
30. Tetsuya Mizuno “Neuron–microglia interactions in neuroinflammation” neuroimmunology. 225–231. 2015.
31. Tulsulkar J, Shah ZA. “Ginkgo biloba prevents transient global ischemia-induced delayed hippocampal neuronal death through antioxidant and anti-inflammatory mechanism.” Neurochem Int. 62(2):189-97. 2013.
32. Zhang YB, Wang X, Meister EA, Gong KR, Yan SC, Lu GW, Ji XM, Shao G. “The effects of CoCl2 on HIF-1α protein under experimental conditions of autoprogressive hypoxia using mouse models.” Int J Mol Sci. 8;15(6):10999-1012. 2014.
33. Zhou J, Schmid T, Frank R, Brüne B.” PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1alpha from pVHL-independent degradation.” J Biol Chem. 2;279(14):13506-13. 2004.
- 국문요약 - LDHB 유전자 삭제에 의한 허혈성 신경세포 사멸 감소 아주대학교 대학원 의생명과학과 분자의학전공 한 송 미 (지도교수: 박 찬 배) 뇌는 체중의 2%를 차지함에도 불구하고 체내 전체 에너지의 약 25%를 소모한다. 이 때문에, 뇌는 특히 허혈상태에 취약하다. 뇌에서 사용되는 에 너지는 대부분 신경세포의 생존이나 활성에 사용된다. 신경세포의 에너지 대사시 포도당보단 젖산이 주로 사용된다고 잘 알려져 있다. 이때, 신경세 포로의 젖산의 이동은 Astrocyte-to-neuron lactate-shuttle (ANLS) 가설에 서 잘 설명된다. 이 연구를 통해 ANLS에서 중요한 효소인 LDHB가 신경세 포에서 어떤 역할을 하는지를 알아보았다. 먼저 LDHB knockout mouse를
만들었고 in vivo와 in vitro 실험에서 LDHB가 없을 때 신경세포가 더 잘 산
다는 것을 보여주었다. In vivo 허혈성 조건에서 LDHB KO 마우스의
posterior communicating artery (PcomA)의 크기가 WT 보다 더 커진 것을 확인하였다. 이 결과는 LDHB KO 마우스에서 PcomA 사이즈의 증가가 신
경세포의 생존에 기여할지도 모른다는 것을 뒷받침한다. 또한 in vitro에서
CoCl2로 허혈성 조건을 만들어 보았을 때 LDHB KO 신경세포에서 VEGFA
발현이 더 증가되어 있음을 확인 할 수 있다. 결론적으로 LDHB KO 신경 세포는 PcomA 사이즈의 증가 및 VEGFA level의 증가를 통해 허혈성 조건
에서 뇌손상에 더 저항성이 있음을 알 수 있었다.
핵심어 : 젖산탈수소효소, 허혈성 스트레스, 신경세포 사멸, 혈관 표피 성장인자