저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Structural and Functional Properties of the
Recombinant anti-KIFC1 Antibodies
Expressed in the Cytosol
by
Youngsil Seo
Major in Molecular Medicine
Department of Biomedical Sciences
The Graduate School, Ajou University
Structural and Functional Properties of the
Recombinant anti-KIFC1 Antibodies
Expressed in the Cytosol
by
Youngsil Seo
A Dissertation Submitted to The Graduate School of
Ajou University in Partial Fulfillment of the Requirements
for the Degree of Ph.D. of Biomedical Sciences
Supervised by
Myung-Hee Kwon, Ph.D.
Major in Molecular Medicine
Department of Biomedical Sciences
The Graduate School, Ajou University
i
-Abstract-
Structural and Functional Properties of the Recombinant anti-KIFC1
Antibodies Expressed in the Cytosol
Intrabodies, antibodies expressed in cells, are widely used in the life science field to regulate the function of intracellular antigens. Most of studies about intrabodies expressed in the cytosol have been performed on single-chain variable fragment (scFv) or single domain antibodies containing few disulfide bonds and simple structure. Because it is known that the disulfide bond is hardly formed in the reducing condition of the cytosol, the antigen-binding site may not be properly formed when the complex structure of immunoglobulin G (IgG) is expressed in the cytosol. In this study, IgG was expressed in the cytosol for the first time to increase the utilization of cytosolically expressed antibody. First, in order to confirm the effect of the variable region on the activity of the cytosolic IgG intrabodies, three anti-KIFC1 (kinesin family C1) IgGs and one anti-nucleic acid IgG that consist of the same human-derived constant regions and different mouse-human-derived variable regions were expressed in the cytosol. Analysis of the cytosolic IgG intrabodies revealed that the antigen-binding activity and the association of heavy and light chains were different according to the variable region sequence. In order to investigate the effect of the constant region folding on the characteristics of the cytosolic IgG intrabodies, cysteine residues in the constant regions were replaced with serine residues to inhibit formation of inter- and intra-chain disulfide bonds. The results suggested that integrity of the constant region affects formation of antigen-binding site of the
ii
variable region. It was also observed that the heavy and light chains were not associated in the absence of intra-chain disulfide bond of constant region. The presence of the disulfide bonds in cytosolic IgG intrabody was confirmed by direct PEGylation of cysteine residues. We also compared the re-folding of fully reduced ER-directed IgG and cytosolic IgG intrabody. It was confirmed that even if the protein had completely the same amino acid sequence, it was folded differently depending on the location of expression. In the case of scFv form of cytosolic 6C407 and 2C281, they retained its antigen-binding activity in the scFv form as well as IgG form. Further, cytosolic 6C407 scFv expression increased multipolar spindle formation and prolonged mitotic duration, suggesting that 6C407 scFv inhibits centrosome clustering function of KIFC1. This study is expected to contribute to the understanding of the structural and functional properties of cytosolically expressed antibodies.
iii
TABLE OF CONTENTS
ABSTRACT ··· i
TABLE OF CONTENTS ··· iii
LIST OF FIGURES ··· v
LIST OF TABLES ··· vii
I. INTRODUCTION ··· 1
II. MATERIALS AND METHODS ··· 11
A. Plasmids ··· 11
B. Purification of proteins ··· 14
C. Preparation of cell lysates··· 15
D. Enzyme-linked immunosorbent assay (ELISA) ··· 16
E. Confocal microscopy ··· 19
F. Immunoblotting ··· 21
G. Immunoprecipitation (IP) ··· 22
H. Measurement of antigen-binding affinity by octet ··· 22
I. Analysis of disulfide bonds by Cys-residue PEGylation ··· 23
J. Analysis of assembly dynamics ··· 24
K. Fluorescence microscopy ··· 24
L. Live cell imaging ··· 25
iv
III. RESULTS ··· 27
A. Expression of IgGs in the cytosol ··· 27
B. Antigen-binding activity of cytosolic IgGs ··· 29
C. Association of heavy and light chains of cytosolic IgGs ··· 33
D. H:L association in the absence of formation of the correct antigen-binding site · 36 E. Effect of structural integrity of the constant region on H:L association and formation of the correct antigen-binding site in cytosolic IgGs ··· 39
F. Importance of intra-chain disulfide bonds in H:L association and antigen- binding site formation of cytosolic IgGs ··· 42
G. Partial formation of intra-chain disulfide bonds in heavy and light chains of cytosolic IgGs ··· 44
H. Differences in assembly of IgG1s expressed in the cytosol or via the ER ··· 48
I. Antigen-binding affinity of purified chimeric anti-KIFC1 IgGs ··· 50
J. Antigen-binding activity of purified anti-KIFC1 scFvs ··· 57
K. Antigen-binding activity of cytosolic scFvs ··· 59
L. Increased multipolar spindle in anti-KIFC1 scFv expressing cells ··· 63
IV. DISCUSSION ··· 69
V. CONCLUSION ··· 73
REFERENCES ··· 74
v
LIST OF FIGURES
Figure 1. Structural features of human IgG1 ··· 2 Figure 2. The major approaches to deliver antibodies into a cell ··· 7 Figure 3. Amino acid sequences of the KIFC1 protein and epitopes recognized
by antibodies ··· 17 Figure 4. Expression of IgGs in the ER or cytosol of HEK293T cells ··· 28 Figure 5. Evaluation of antigen-binding activity of cytosolic IgGs ··· 31 Figure 6. Confocal microscopic analysis to confirm antigen-binding site of the
cytosolic IgGs··· 32 Figure 7. Association of heavy and light chains of cytosolic IgGs ··· 34 Figure 8. Evaluation of H:L association of cytosolic IgGs ··· 35 Figure 9. Association of heavy and light chains without formation of the correct
antigen-binding site ··· 37 Figure 10. Effect of the absence of disulfide bonds in constant regions on H:L
association and correct folding of antigen-binding site in cytosolic IgGs ··· 40 Figure 11. Effect of intrachain disulfide bonds on H:L association and antigen-binding
site formation of cytosolic IgGs ··· 43 Figure 12. Absence of spontaneous ex vivo non-enzymatic disulfide bonds formation
in cytosolic IgGs ··· 46 Figure 13. Presence of disulfide bonds in the cytosolic IgG ··· 47 Figure 14. Comparison of assembly patterns of IgG expressed in reduced cytosol and
vi
through oxidative environment of ER ··· 49 Figure 15. Purification of chimeric anti-KIFC1 IgGs ··· 52 Figure 16. Measurement of antigen-binding affinity of purified chimeric anti-KIFC1
IgGs ··· 53 Figure 17. Measurement of antigen-binding affinity of purified anti-KIFC1 scFvs ··· 54 Figure 18. Antigen-binding activity of purified anti-KIFC1 scFvs ··· 58 Figure 19. Confocal microscopic analysis to confirm antigen-binding activity of
cytosolic scFvs ··· 60 Figure 20. Evaluation of Antigen-binding activity of cytosolic anti-KIFC1 scFvs ··· 61 Figure 21. Investigation of binding activity of cytosolic scFvs to endogenous KIFC1
by IP ··· 62 Figure 22. Increased multipolar spindle formation in anti-KIFC1 scFv expressing cells · 66 Figure 23. Prolonged mitotic duration of MDA-MB-231 cells expressing cytosolic
6C407 scFv ··· 67 Figure 24. Delayed meta-to-anaphase transition upon 6C407 scFv expression
vii
LIST OF TABLES
Table 1. Amino acid sequence of anti-KIFC1 antibody variable regions ··· 13 Table 2. Sequence of siRNA ··· 17 Table 3. Antigen-binding affinity of anti-KIFC1 antibodies ··· 56
1
I. INTRODUCTION
Antibodies are Y-shaped proteins in which two heavy chains and two light chains are linked by disulfide bonds (Figure 1). Each chain is composed of immunoglobulin domains, with variable and constant regions (Stanfield and Wilson, 2014). The constant region determines the isotype of the antibody, is recognized by the Fc receptor on the surface of immune cells and plays a role in mediating various immune responses. The variable region is the site that shows specificity for an antigen. The variable domain consists of four framework regions and three complementarity determining regions (CDRs). The framework region is the backbone of the antibody and the CDR shows specificity for the antigen because of its high variability. Due to differences in the amino acid sequence of the complementarity determining region, each antibodies have its own characteristics (Glockshuber et al., 1990).
2
Figure 1. Structural features of human IgG1. IgG1 protein contains 4 inter-chain disulfide bonds and 12 intra-chain disulfide bonds. VH: variable heavy chain, VL: variable light chain, CH: constant heavy chain, CL: constant light chain, S-S: disulfide bond.
3
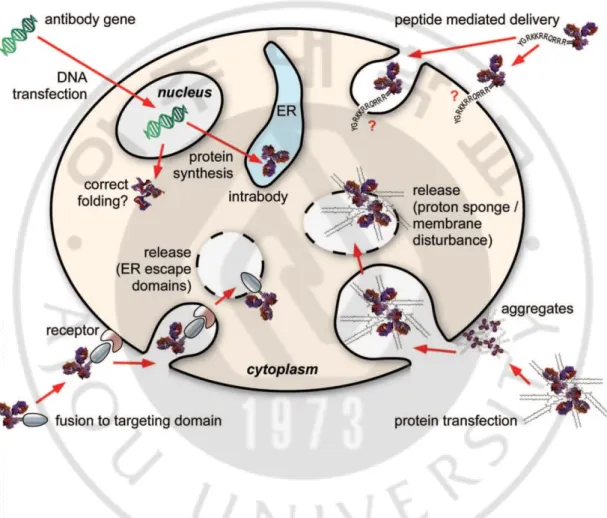
The ability of antibodies to show high specificity for a protein can be a good tool to enhance the understanding and regulation of phenomena that occur inside living cells (Accardi et al., 2014). Because half of the human proteome is present in the cell, a variety of proteins can be targets of the antibody if the antibody can act inside the cell. Inside cells, single antibodies can be used to target multiple protein isoforms and to knockdown protein functions by targeting splice variants or even post-translational modifications (Liu et al., 2015; Marschall et al., 2015). These functions cannot be obtained using DNA or RNA. Intracellular antibodies also enable live cell imaging allowing tracking of the target protein in living cells (Marschall et al., 2011; Rinaldi et al., 2013). In order to control the function of the intracellular molecule, a small molecule inhibitor is used, which is small and can easily enter the cell. However, using small-sized small molecule materials to control the function of large molecules such as proteins is not suitable because of the large number of unexpected off-target effects. In contrast, the antibody exhibits high antigen specificity for the protein, so it is suitable for targeting proteins because the off-target effect is low.
To make antibodies function within cells, antibodies should be localized inside cells. However, large size of antibody makes them difficult to enter the cell via passive transport. Many researchers have tried various methods to effectively deliver antibodies into cells (Ye et al., 2002). Initially, microinjection method was used whereby a needle was used to deliver the antibody into cells, where they successfully bound to the target in the cells (Blose et al., 1984; Ozawa et al., 1985; Fu et al., 2014). However, this method induced a high rate of apoptosis and is difficult to apply to large scale experiments due to requirement of injection
4
of antibody into individual cells. Liposome-mediated delivery, which is easier to manipulate than microinjection, has been widely used to deliver proteins as well as nucleic acids. Protein transfection (profection) reagents increase the permeability of the cell membrane and transfer the protein from the outside into the cell. According to a paper by Marschall and colleagues, the protein delivery efficiency of these reagents was high, but the rate at which the protein delivered into the cells escaped the endosome and released into the cytosol was significantly low. They reported that the recombinant fusion protein of cre recombinase and human IgG1 Fc was transferred to the reporter cell and quantified the cytosolic delivery efficiency of the fusion protein by measuring the GFP reporter signal generated by cre recombinase activity. Four protein transfection reagents were used to transfer the fusion protein to the cells under the conditions suggested by the manufacturer and observed for up to 96 hours. The percentage of cells that were reporter gene-activated was only a single digit. Endosomal release is a limiting factor in observing the function of antibodies delivered into cells. Kim and colleagues devised a method to quantitatively measure the release of antibodies into cytosol (Kim et al., 2015). In this study, the ratio of antibody escaped from the endosome (endosomal-escaping efficiency) among the TMab4 antibodies that enter the cell by itself was measured quantitatively by the split-GFP complementation assay. As a result, only 1.3 to 4.3% of the antibodies entering the cell were released into the cytosol, indicating that a small amount of endosomal released antibody may act as a limiting factor in the function of the intracellular antibody. Compared to lipid-based profiling reagents, the proportion of proteins released into the cytosol was higher when electroporation was performed (Marschall et al., 2014b). A
5
number of studies have also been conducted to apply the method of electroporation, which temporarily creates small holes on the cell membrane by electric shock, to protein delivery (Chakrabarti et al., 1989; Graziadei et al., 1991; Freund et al., 2013; Clift et al., 2017). However, in the case of proteins compared to DNA or siRNA, the process of finding optimal conditions for effective delivery is difficult, since the structure and charges vary from protein to protein (Brees and Fransen, 2014). In addition to delivering the purified antibody by physicochemical methods, there is a method of tagging a peptide having cell penetration activity to the antibody (Tashima, 2017). Cell-penetrating peptides (CPPs) have been extensively studied because they are small in size and are easily transferred into cells with little effect on the function of the antibody (Bechara and Sagan, 2013). Among protein-derived CPP, Tat peptide (Tat 48-60, GRKKRRQRRRPPQ) originated from the transcriptional transactivator of the human immunodeficiency virus is the most widely used CPP (Vives et al., 1997). Tat peptide is positively charged due to basic residues such as lysine and arginine. Cell penetration of Tat peptide can be achieved by interacting with negatively charged cell membrane components. Among synthetic peptides, positively charged polyarginine peptides composed of 6 to 12 arginine residues are widely used (Futaki et al., 2001; Wang et al., 2016). The mechanism of cell penetration differs dependent on the peptide. Some CPPs enter the cells and are trapped in endosomes and then degraded by lysosomes. It has been found that CPP introduced into cells by endocytosis escapes endosomes and is released into cytoplasm by avoiding lysosomal degradation (Madani et al., 2011). In order to
6
introduce the antibody into the cell using CPP, an additional cloning step is essential to label CPP to the antibody.
Instead of a purified antibody protein, an antibody gene can be delivered into the cell, allowing the antibody to be expressed and function within the cell (Persic et al., 1997). Intrabody is an antibody that is expressed in a cell and functions in the same cell in which the antibody is made. Introduction of the gene of the antibody in the cell, followed by expression is the way to accumulate surplus antibody in the cell without the purification step. Intracellular localization of the intrabody can be controlled by the localization signal, so that it can be accurately transferred to the nucleus, mitochondria, and ER (Kirschning et al., 2010; Marschall et al., 2014a; Lee et al., 2016).
7
8
In natural state, antibodies are proteins that function after being secreted out of the cell (Braakman and Bulleid, 2011). Therefore, the intracellular antibody is not optimized to work in the intracellular environment, so even if it functions outside the cell, it may not function inside the cell (Glockshuber et al., 1990). This is a characteristic that varies from one antibody to another and cannot be known until directly expressed and confirmed (Tanaka and Rabbitts, 2008).
The variable region, which determines the antigen-binding site, is linked by a disulfide bond between the heavy and light chains (Feige et al., 2010). The disulfide bond of an antibody is formed in the endoplasmic reticulum (ER) during the secretory pathway (Feige et al., 2010). Protein disulfide isomerase (PDI), which is present in the ER, is self-reduced and oxidizes the cysteine residue in the polypeptide chain entering the ER to form a disulfide bond (Hamers-Casterman et al., 1993; Braakman and Bulleid, 2011). Unlike in the ER, there is no PDI in the cytosol (Cumming et al., 2004; Appenzeller-Herzog and Ellgaard, 2008). In addition, glutathione reductase and thioredoxin reductase that are present in the cytosol maintain the reducing environment continuously (Sevier and Kaiser, 2002). Therefore, it is known that disulfide bonds are rarely formed in the proteins expressed in the cytosol (Biocca et al., 1995; Frisch et al., 1996). When the antibody is expressed in the cytosol, it folds in a different environment from the conventional antibody. Therefore, in order to utilize an antibody in the cell, it should be verified whether the antigen-binding activity is maintained even under the reducing condition of the cytosol. In order to make the antibody function
9
within the cell, studies should be conducted to identify differences in the structural or functional properties of the antibody depending on location of antibody generation.
In this study, I analyzed structural and functional characteristics of antibodies purified from culture supernatant or cytosolically expressed in the format of IgG or scFv. Three kinds of antibodies are specific to KIFC1 (kinesin family member C1) and one is an anti-DNA antibody (3D8).
KIFC1, a minus directed kinesin motor protein, has been known to be essential for mitotic spindle formation during mitosis (Kwon et al., 2008). Cancer cells harboring extra-centrosomes are highly dependent on the centrosome clustering activity of KIFC1 for bipolar division, leading to elevated expression of KIFC1 in breast and ovarian cancer (Pawar et al., 2014; Pannu et al., 2015; Mittal et al., 2016). It is well known that depletion of KIFC1 in cancer cells with supernumerary centrosomes causes multipolar spindle formation, resulting in abnormal division (Kwon et al., 2008). Recent studies revealed that KIFC1 is implicated in acentrosomal spindle organization (Kleylein-Sohn et al., 2012) and in direct interaction with CEP215 (Chavali et al., 2016), supporting the important role of KIFC1 in the linking of spindle pole and centrosomes. As KIFC1 is considered an attractive anti-cancer target molecule (Li et al., 2015; Myers and Collins, 2016), KIFC1 inhibitors have been developed for the functional knockdown of KIFC1 in cancer (Watts et al., 2013; Wu et al., 2013; Zhang et al., 2016). As KIFC1 is located in the nucleus of the cell, KIFC1 can only interact with cytosolic anti-KIFC1 antibody after the nuclear membrane has broken down in mitosis (Cai
10
et al., 2009). 3D8 is an auto-antibody isolated from an autoimmune-prone MRL-lpr/lpr mouse and binds to both DNA and RNA without sequence specificity.
Most of the experiments were carried out after expressing the antibodies in the mammalian cells and then confirming the expression level of the antibodies by immunoblotting. In the case of cytosolic antibodies, antigen-binding activity and assembly between heavy and light chains was confirmed by various methods such as cell lysate enzyme-linked immunosorbent assay (ELISA), immunoprecipitation (IP), and confocal microscopy. Antigen-binding affinity of the purified antibody was measured by octet and antigen-binding activity was demonstrated by ELISA or confocal microscopy. This study suggests that structural and functional characteristics of cytosol-expressed antibody compared with ER-directed antibody may be a basis for widening intrabody application.
11
II. MATERIALS AND METHODS
A. Plasmids
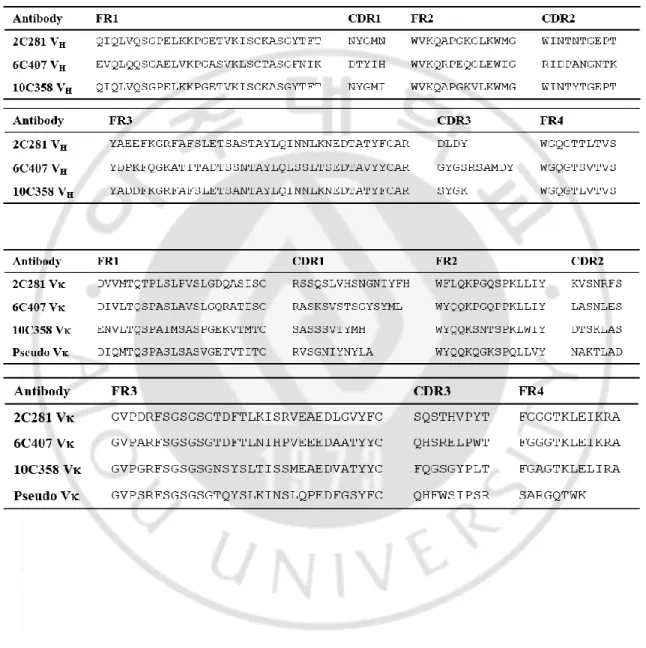
KV10 vector was used to express the protein in mammalian cells. Since two CMV promoters are contained in the KV10 vector, two proteins can be expressed simultaneously. For the expression of IgG, variable heavy chain (VH) and constant gamma chain 1-3 (Cγ1, Cγ2, and Cγ3) genes were inserted into one promoter to express heavy chain, while variable light chain (VL) and constant kappa chain (Cκ) genes were inserted into the other promoter to express light chain. A variable region gene derived from a mouse was inserted upstream of the human IgG1 constant region to be expressed as chimeric IgG. The DNA sequence and amino acid sequence of the variable region gene of the antibody are registered in GenBank and the amino acid sequence is aligned in Table 1 (3D8 VH, GenBank accession number AAF79128; 3D8 VL, AAF79129; 2C281 VH, MH638364; 2C281 VL, MH638365; 6C407 VH, MH638366; 6C407 VL, MH638367; 10C358 VH, MH638368; 10C358 VL, MH638369).
Since the KV10 vector contains different restriction enzymes for each region, it is possible to insert or replace a specific gene at a desired site. For secretion expression of the antibody, the VH region is located between the MluI/NheI and the Cγ1-3 regions between the
NheI/BamHI restriction enzyme sites. The Vκ region and the Cκ region are located between
DraIII/BsiWI and BsiWI/EcoRI, respectively. There is a leader sequence at the amino terminus of each variable region. The leader sequence derived from the VH3 gene family for VH and VκI for Vκ is located between MfeI/MluI and BglII/DraIII, respectively. When IgG was expressed in the cytosol, the VH and Vκ gene fragments not containing the leader
12
sequence were prepared and inserted between the restriction sites of MfeI/NheI and BglII/BsiWI, respectively.
For the expression of mutant ΔLd-IgG *#H*#L, all cysteine residues in IgG constant regions were replaced with serine to prevent intra- and inter-chain disulfide bond formation. Ig-whole constant (Cw), consisting of only constant region without variable region, was constructed by synthesizing Cγ1-3 and Cκ gene with WT and mutant and inserting into KV10 vector. For secretion expression of Ig-Cw, heavy chain was inserted between MluI/BamHI and light chain was inserted between DraIII/EcoRI restriction site. For the cytosolic expression, heavy chain was inserted between MfeI/BamHI and light chain was inserted between BglII/EcoRI. ΔLd-3D8 IgG/H-HA and ΔLd-3D8 IgG/L-Flag were constructed by inserting the Cγ1-3-HA gene between NheI/BamHI and the Cκ-Flag gene between
BsiWI/EcoRI.
In order to express scFv, the VH and VL genes of the antibody were linked by a (Gly4Ser)3 linker through PCR. To purify the scFv in E. coli, the scFv gene was inserted between the XmaI/NcoI restriction sites in the pIg20 E. coli expression vector. In order to express scFv in mammalian cells, scFv-HA and scFv-myc genes were prepared and inserted between the MfeI/NheI for cytosol expression and MluI/NheI for expression through ER-directed pathway of KV10 vector. The stop codon was inserted behind the tag to prevent additional translation.
13
14
B. Purification of proteins
Chimeric anti-KIFC1 IgG proteins were expressed in the HEK293F cells transfected with KV10 Ld-anti-KIFC1 IgG plasmid. HEK293F cells were cultured in serum-free Freestyle medium (Gibco; cat# 12338018) in 500 ml flask (Corning; cat# 431145) at 8% CO2 and 37C with shaking at 130 rpm. Cells were seeded 1×106 cells/ml density in 100 ml medium 24 h prior to transfection. Plasmids DNA (200 μg) encoding antibody gene was mixed with PEI (Polyscience; cat# 23966-2, 400 μg) in 10 ml of medium and incubated for 10 min at RT. The mixture was inoculated into 100 ml of HEK293F cells culture and incubated for 7 days. The supernatant was harvested by centrifugation and was passed through Protein-A affinity chromatography column (GE Healthcare; cat# 17-1279-02) to purify IgG proteins. After washing the column with PBST, proteins were eluted with 0.1 M of glycine (pH 3.0). The eluate was neutralized with 1 M Tris (pH 9.5) and then concentrated by Vivaspin (molecular cut-off 50,000 kDa, Sartorius; cat# VS2032). The buffer was changed into PBS.
Anti-3D8 idiotypic O2F3 mouse IgM, which recognizes the conformation of the antigen-binding site composed of VH and VL, was purified from the culture supernatant of O2F3 hybridoma. O2F3 hybridoma cells were cultured in RPMI 1640 media supplemented with 10% FBS at 5% CO2 and 37°C. O2F3 IgM in culture supernatant was purified by affinity chromatography using agarose-goat anti-mouse IgM / μ chain-specific resin (Sigma-Aldrich; cat# A4540).
Purification of the scFv protein was done in E. coli. BL21 (DE3) pLysE (Novagen) were transformed with pIg20-anti-KIFC1 scFv plasmid and grown in 1 L of Luria Bertani medium with 100 μg/ml of ampicillin and 25 μg/ml of chloramphenicol at 37°C, until absorbance
15
reaches 0.6 at 600 nm. Expression of scFv protein was induced by addition of 0.5 mM of isopropyl 1-thio-β-D-galactopyranoside. After growing cells at 23°C for 18 h, cells were collected by centrifugation at 8000 rpm for 30 min at 4°C. The supernatant was passed through IgG-sepharose affinity column (GE healthcare; cat# 17-1279-02) and scFv protein was eluted by 0.1 M acetic acid (pH 3.4). Eluted solution was concentrated in Vivaspin (molecular cut-off 10,000 kDa, Sartorius; cat# VS2002) by centrifugation and the buffer was changed to PBS.
C. Preparation of cell lysates
HEK293T cells were seeded 1×107 cells in 150 mm dish 24 h prior to transfection. Plasmid DNA (20 μg) and PEI (80 μg) reagent were mixed in 10 ml of media and incubated at RT for 10 min. The mixture was inoculated into cells in 10 ml of Opti-MEM (Gibco; cat# 31985-070) at 37C for 36 h. If it is needed, 50 mM of N-ethyleneamine (NEM, ThermoFisher; cat# 23030) was treated and incubated at 37C for 15 min. After collecting cells by pipetting, cells were washed with PBS for three times and resuspended with ice-cold PBS containing protease inhibitor cocktail (Roche; cat# 11697498001) or 100 mM of NEM. Cell were lysed by sonication at 30% amplitude by three pulses for 10 sec (Epishear). The lysed solution was centrifuged at 16,000 × g at 4C for 10 min and the supernatant was collected.
16
D. Enzyme-linked immunosorbent assay (ELISA)
Cell lysate ELISA for the analysis of antigen-binding activity
To assess the antigen-binding activity of anti-KIFC1 antibodies (2C281, 6C407 and 10C358), lysates of transfected HEK293T cells were incubated in a 96-well plate coated with synthetic peptide antigens (1 μg/ml, Figure 3). Synthetic peptide antigens for 2C281 consisted of amino acids 70−81 (PSLTTVPQTQGQ, KIFC1 peptide #1) of KIFC1. Peptide antigens for 6C407 and 10C358 consisted of amino acids 43−54 (EDGLEPEKKRTR, KIFC1 peptide #2) and 99−110 (IATGLKNQKPVP, KIFC1 peptide #3) of KIFC1. Bound anti-KIFC1 IgGs were detected with alkaline phosphatase (AP)-conjugated goat anti-human IgG/Fc antibody (Sigma-Aldrich; cat# A9544). Bound anti-KIFC1 scFvs were detected with rabbit anti-HA antibody (Abcam; cat# ab9110), followed by AP-conjugated goat anti-rabbit IgG/Fc (Thermo; cat# 31341). Where necessary, lysates of transfected HEK293T cells were prepared in the presence of 100 mM NEM. ρ-nitrophenyl phosphate (Sigma-Aldrich; cat# N2765) solution (1 mg/ml in 0.1 M glycine, 1 mM ZnCl2 and 1 mM MgCl2, pH 10.3) was added to each well and the absorbance at 405 nm was measured using a microplate reader (Molecular Devices).
17
Figure 3. Amino acid sequences of the KIFC1 protein and epitopes recognized by antibodies. The epitope sequences recognized by 6C407, 2C281, and 10C358 antibodies are indicated by red, blue, and green amino acids, respectively.
18
Cell lysate ELISA for the analysis of H:L association
To assess the association of heavy and light chains in cells, two different ELISA protocols were used. In the first method, wells of a 96-well polystyrene plate were coated with 100 μl (2 μg/ml) of goat anti-human IgG/Fc antibody (Abcam; cat# ab97221) for 1 h at RT, washed three times with TBST, and blocked with 3% bovine serum albumin (BSA) for 1 h at RT. Wells were subsequently incubated with lysates of transfected cells (100 μl) for 1 h at RT, rabbit anti-human C antibody (Abcam; cat# ab125919), and AP-conjugated goat anti-rabbit IgG/Fc-specific antibody (Pierce; cat# 31341). Each incubation step was followed by washing three times with TBST. Otherwise, the plate coated with 100 μl (2 μg/ml) of rabbit anti-human C antibody was incubated with lysates of transfected cells, followed by AP-conjugated goat anti-human IgG/Fc-specific antibody (Sigma-Aldrich; cat# A9544). Polyclonal human IgG (Sigma-Aldrich; cat# I8640) was used as a positive control.
ELISA for the analysis of antigen-binding activity of purified scFvs.
The antigen-binding activity of anti-KIFC1 scFvs (2C281 and 6C407) was assessed by ELISA using synthetic peptides as antigens. Purified anti-KIFC1 scFvs (1 μM) were treated into the peptide antigens (1 μM)-coated 96-well plate and incubated at RT for 1 hr. Peptide KIFC1 1-50 was used as the 6C407 antigen and KIFC1 51-100 was used as the 2C281 antigen. Bound scFvs were detected by rabbit IgG and AP-conjugated goat anti-rabbit IgG. DTT was treated to scFv protein at a 55 mM concentration. HW6 scFv was used as a negative control.
19
E. Confocal microscopy
For analysis of colocalization of anti-KIFC1 IgG intrabodies and cellular KIFC1 molecules, HeLa cells stably expressing GFP-KIFC1 were seeded on cover slips and transfected with plasmids encoding 2C281, 6C407, or KV10Ld-10C358. At 24 h after transfection, cells were synchronized at mitosis by double thymidine block. To arrest most cells in G1/S, cells were treated with 2 mM of thymidine for 18 h, washed with PBS three times, and placed in fresh medium for 7 h. After that, to arrest cells at the G2/M phase border, cells were incubated with 9 μM of CDK inhibitor RO3306 (Sigma-Aldrich; cat# SML0569) for 2 h, and then placed in fresh medium for 30 min, fixed, and permeabilized. Cells were incubated with goat anti-human IgG/Fc-specific antibody (Abcam cat# ab97221) followed by TRITC-conjugated rabbit anti-goat IgG (Abcam; cat# ab50623). Finally, cell nuclei were stained with Hoechst 33342 (Vector Laboratories) for 30 min at RT. Thereafter, cells on coverslips were mounted with Vectashield mounting medium (Vector Laboratories). Images were obtained using a laser scanning confocal fluorescence microscope (Carl Zeiss, LSM710).
To analyze colocalization of cytosolically expressed 3D8 IgG and O2F3 antibodies (mouse IgM specific for conformational variable regions of 3D8 (Kwon et al., 2002)), HEK293T cells were seeded on poly L lysine-coated glass coverslips in 24-well plates at a density of 4×104 cells/well, prior to transfection with KV10Ld-3D8 plasmid using Lipofectamine 2000 reagent (Life Technologies). At 24 h after transfection, cells were washed three times with ice-cold PBS (pH 7.2), and fixed with 4% paraformaldehyde in PBS for 10 min at 4C. After washing cells three times with PBS, cell membranes were
20
permeabilized with P buffer consisting of 1% BSA, 0.1% saponin, and 0.1% sodium azide in PBS for 10 min at RT. Cells were incubated overnight at 4C with O2F3 antibody, and then with an Alexa Fluor 647-conjugated rat anti-mouse IgM/µ chain-specific antibody (BioLegend; cat# 406526). After each incubation for 1 h at 4C, cells were washed three times with ice-cold PBS.
For the in vitro staining using purified anti-KIFC1 scFvs, HeLa cells expressing KIFC1-GFP were fixed with 4% paraformaldehyde in PBS (pH 7.4) following cell membrane permeation with P buffer in the dark. Purified scFvs (10 μM) were treated and incubated for 1h at RT. Rabbit IgG (Sigma; cat# I5006) and TRITC conjugated goat anti-rabbit IgG (Sigma; cat# T6778) were treated to detect purified scFvs tagged with protein A.
To analyze co-localization of cytosolic anti-KIFC1 scFv and endogenous KIFC1, scFvs labeled with HA tag was expressed in the cytosol of HeLa GFP-KIFC1 cells. Cells were synchronized at mitosis by double thymidine block, fixed with 4% paraformaldehyde in PBS and membrane was permeabilized with P buffer. Cytosolic anti-KIFC1 scFv was detected by mouse anti-HA antibody (Millipore; cat# 05-904) and TRITC conjugated goat anti-mouse IgG (Abcam; cat# ab47832). After nucleus staining and mounting, stained cells were observed with a confocal microscope.
21
F. Immunoblotting
For SDS-PAGE analysis under non-reducing conditions, samples were diluted 1:1 in 2 Laemmli sample buffer (Bio-Rad). For reducing conditions, samples were diluted 1:1 in 2 Laemmli sample buffer supplemented with 2.5% 2-mercaptethanol or 100 mM DTT. In both cases, samples were heated at 100C for 10 min, then loaded on 8%, 10%, or 4−20% acrylamide gels. Following electrophoresis, resolved proteins were transferred onto polyvinylidene fluoride membranes. Membranes were rinsed with PBS, then blocked with 5% milk (w/v) in TRIS-buffered saline (TBS, 50 mM TRIS-Cl, 50 mM NaCl, pH 7.2) containing 0.05% Tween-20 (TBST) at RT for 2 h. Membranes containing Ig proteins were probed with primary goat anti-human IgG/Fc antibody (Pierce; cat# 31125) plus rabbit anti-human Ig chain antibody (Abcam; cat# ab134083) in 5% milk-TBST at 4C overnight. After washing three times with PBS containing 0.05% Tween-20 (PBST), membranes were incubated with secondary horseradish peroxidase (HRP)-conjugated anti-goat IgG (Invitrogen; cat# 81-1620) plus HRP-conjugated anti-rabbit IgG (Invitrogen; cat# 81-6120) for 1 h. After washing five times with TBST, protein signals on membranes were visualized with an ECL kit (GE Healthcare; cat# RPN2106). Where necessary, membranes were probed with primary mouse anti-HA antibody (Millipore; cat# 05-904) plus mouse Flag antibody (Sigma-Aldrich; cat# F3165), followed by HRP-conjugated horse anti-mouse IgG (Cell Signaling; cat# 7076).
22
G. Immunoprecipitation (IP)
Aliquots (500 μg) of cell lysate was subjected to IP with Protein A/G coupled to resin (ThermoFisher; cat# 26146) according to the manufacturer’s instructions. After washing the resin, immunoprecipitated heavy chain of antibody were eluted and resolved by SDS-PAGE. Co-immunoprecipitated light chain was detected by immunoblotting with rabbit anti-human Ig chain antibody (Abcam; cat# ab134083).
For the IP of myc-tagged anti-KIFC1 scFvs, mouse anti-myc antibody (IGTHERAPY; cat# IG-A200302) was coupled to agarose resin (ThermoFisher; cat# 26146). Lysates of HeLa cells expressing scFvs (500 μg) were incubated with the anti-myc IgG coupled resin at 4°C overnight. Bound scFvs were eluted according to the manufacturer’s instructions. Eluted proteins were separated by SDS-PAGE and immunoblotting were performed with rabbit anti-KIFC1 antibody (abcam; cat# ab172620) and mouse anti-myc antibody.
H. Measurement of antigen-binding affinity by octet
Affinities of purified anti-KIFC1 IgGs and scFvs against peptide antigen were measured by octet systems (Fortebio). Biotin-conjugated synthetic KIFC1 peptides (KIFC1 1-50 for 6C407 and KIFC1 51-100 for 2C281) were immobilized to streptavidin coated biosensor in an optimized concentration condition (1 μg/ml of KIFC1 1-50 for 6C407 IgG, 1 μg/ml of KIFC1 1-50 for 6C407 IgG, 0.25 μg/ml of KIFC1 51-100 for 2C281 scFv, and 0.5 μg/ml of KIFC1 51-100 for 2C281 IgG). Proteins that were serially diluted 2-fold starting at 200 nM concentration were analyzed for more than 4 concentrations. The association and dissociation of the antibody was measured for 300 sec or more. Fitting curves and dissociation constant (KD) were obtained using the Fortebio Data analysis program provided by the manufacturer.
23
I. Analysis of disulfide bonds by Cys-residue PEGylation
IgG was expressed in the cytosol of HEK293T by transient expression. Cells were transfected with KV10ΔLd-3D8 IgG/H-HA plasmid DNA using PEI and incubated for 36 hrs. Before harvesting the cells, NEM was treated to the medium at a concentration of 50 mM to block the oxidation of free thiol. Collected cells were washed twice with PBS after centrifugation in 3000 rpm for 10 min at 4°C. The cell pellet was resuspended in PBS containing 100 mM NEM and lysed by sonication at 30% amplitude three times for 10 seconds. The supernatant was collected by centrifugation with 13000 rpm for 10 min at 4°C and concentration of protein was measured by BCA assay. To remove the NEM in the supernatant, cell lysate was passed through desalting column (7 kDa molecular weight cut off, Thermo; cat#89882). For the reduction of disulfide bonds in the proteins, 5 mM of TRIS-(2-carboxyethyl) phosphine (TCEP) in the HEPES (pH 7.0) was treated to the NEM-removed cell lysate and incubated for 1 h at 37°C. Then, a linear monofunctional methyl ether poly-ethylene glycol with a reactive maleimide group (mPEG-MAL, 2 kDa-size, Creative PEGWorks; cat# PSB-235) constituted in PBS (pH 7.4) was treated at a final concentration of 10 mM and incubated for 1 h at 37°C. PEGylated proteins were mixed with SDS sample buffer in reducing condition and separated by 8% SDS-PAGE gel. Heavy chain of PEGylated IgG was detected by mouse anti-HA tag antibody (Millipore; Cat#. 05-904) and HRP conjugated horse anti-mouse IgG (H+L) antibody (Cell Signaling; Cat# 7076). Total PEGylated proteins were detected by rabbit anti-PEG antibody (abcam; Cat# Ab51257) and HRP conjugated goat anti-rabbit IgG (Zymed; Cat# 81-6120).
24
J. Analysis of assembly dynamics
HEK293T cells were transfected with KV10Ld-3D8 IgG or KV10Ld-IgG at 37C for 24 h. Aliquots of transfectant lysates were treated with 100 mM DTT for 30 min, then passed through a desalting column (ThermoFisher; cat# 89882) equilibrated with PBS, followed by micro-dialysis against PBS using a micro-dialyzer (ThermoFisher; cat# 88260) at 4C for 24 h. Proteins were subjected to immunoblotting analysis in non-reducing and denaturing conditions using anti-IgG/Fc antibody. To validate the complete removal of DTT in the above column and dialysis steps, 100 mM DTT in PBS was passed through the desalting column then subjected to dialysis against PBS, and the concentration of DTT was determined using a free thiol detection kit (Abcam; cat# ab112158).
K. Fluorescence microscopy
To observe multipolar spindle formation, anti-KIFC1 scFv was expressed in the cytosol of HeLa, MDA-MB-231, and RPE-1 cells by DNA transfection. The expression level of KIFC1 was knocked down using siRNA as a control for the inhibition of KIFC1 function. The sequences of siRNA are shown in Table 2. After 24 h, the cells were treated with 100 μM of thymidine for 24 h and released for 9 h. MG132 was treated for 1 h at a final concentration of 10 μM. After fixation and permeation of the cells, the centrosomes were stained with mouse γ-tubulin IgG (Sigma; cat# T6557) and Alexa488-labeled rabbit anti-mouse IgG (Invitrogen; cat# A11059). Stained cells were observed under a fluorescence microscope (Zeiss, Axiovert 200M). The number of cells containing the multipolar spindle was counted at least three times.
25
L. Live cell imaging
For the analysis of the effect of cytosolic anti-KIFC1 scFvs expression on the mitosis duration, live cell imaging was performed. Transfected HeLa cells were placed in a stage-top incubation chamber and monitored every 3 minutes for 48 h by inverted fluorescence time-lapse microscope (Nikon, Ti-E). The mitotic duration was measured from cell round-up to anaphase onset. Mitotic duration refers to the time taken from when the cell begins to rise until it begins to divide.
M. Analysis of meta-to-anaphase delay
The meta-to-anaphase delay of the mitotic cell was confirmed by immunoblotting when scFv was expressed in the cytosol. Plasmids encoding the scFv gene are transfected into HeLa cells and treated with 100 μM of thymidine. After 24 h, the medium was replaced with thymidine-free medium and cells were harvested every hour from 8 h later. Harvested cells were mixed with 2xSDS buffer and directly lysed at 100°C for 5 min. SDS-PAGE and immunoblotting were performed to detect phospho-histone H3 (Cell signaling; cat# 9706), cyclin B1 (Santa Cruz; cat# SC-245), and GAPDH (Cell signaling; cat# 2118) levels in the cell lysate.
26
Table 2. Sequence of siRNA
Oligo sequence 5’-3’ si-control UUCUCCGAACGUGUCACGUTT si-KIFC1 UAACUGACCCUUUAAGUCCUU AGUGUUGUGCGCUCUGUCCUU GACACAAGCACGCAAGUUCUU UGGUCCAACGUUUGAGUCCUU
27
III. RESULTS
A. Expression of IgGs in the cytosol
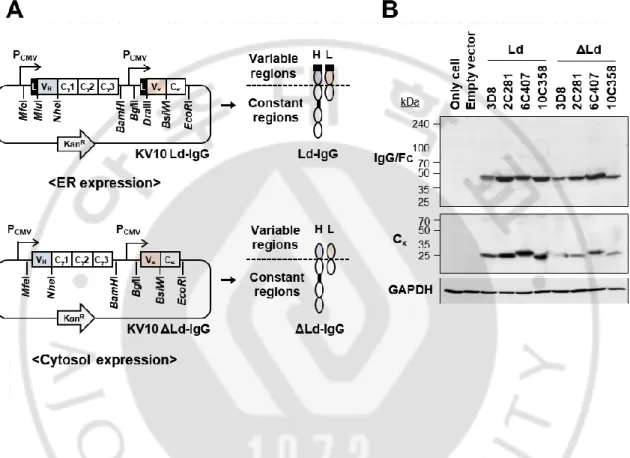
To investigate whether the characteristics of the antibody expressed in the cytosol are determined according to the variable region sequence, chimeric IgG was constructed by fusing four variable regions of mouse-derived antibodies (3 anti-KIFC1 antibodies and an anti-nucleic acid antibody) with the human IgG1 constant region (Figure 4A). A heavy chain and a light chain gene of IgG were inserted into a vector containing two promoters so that each chain could be expressed simultaneously. At the N-terminus of each chain, there is a leader sequence that transfers proteins to the ER when protein translation begins. The IgG expressed in ER was designated Ld. Cytosolic expression was achieved by removing the leader sequence at the N-terminus of the heavy and light chains, and the antibody expressed in the cytosol was labeled as ΔLd. IgGs were expressed in the ER or cytosol of HEK293T cells and expression level was confirmed by immunoblotting (Figure 4B). In the results, all heavy chains were expressed at similar levels regardless of the leader sequence. Light chain of 3D8 and 10C358 showed slightly lower cytosolic expression levels than those expressed in ER (Figure 4B).
28
Figure 4. Expression of IgGs in the ER or cytosol of HEK293T cells. (A) Schematic diagram of plasmids construction to express chimeric IgGs in the ER (Ld) or cytosol (ΔLd). Heavy and light chains of IgG were expressed simultaneously using dual promoter vector. (B) Immunoblotting for detection of heavy and light chains in lysates of HEK293T cells. An anti-DNA antibody (3D8) and three anti-KIFC1 antibodies (2C281, 6C407, and 10C358) were expressed in the ER or cytosol of HEK293T cells. The expressions of heavy and light chains were detected by goat anti-human IgG (Fc specific) antibody and goat anti-human kappa chain antibody followed by HRP-conjugated rabbit anti-goat IgG (H+L).
29
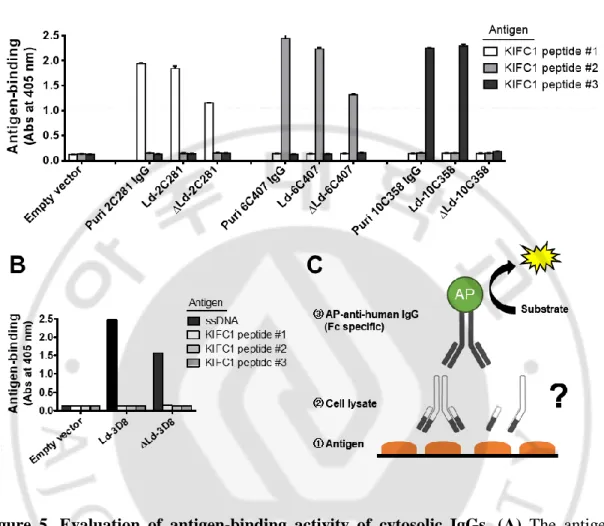
B. Antigen-binding activity of cytosolic IgGs
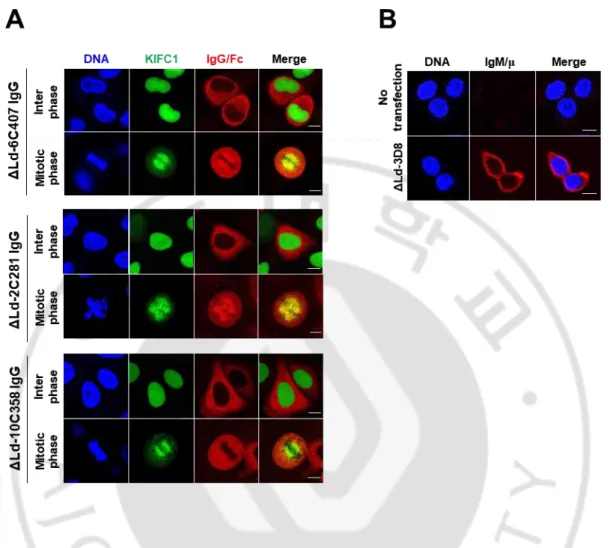
Antigen-binding activity of cytosolic IgGs was confirmed by ELISA and confocal microscopy. In ELISA, cell lysates prepared by disrupting HEK293T cells expressing cytosolic IgG were used. The ELISA plate coated with KIFC1 peptide antigens for anti-KIFC1 antibodies or single-strand DNA antigen for 3D8. All antibodies, except 10C358, expressed in the cytosol showed antigen-binding activity. All the antibodies expressed in ER had antigen-binding activity (Figure 5). This result was confirmed again by confocal microscopy (Figure 6). To confirm whether the cytosolic anti-KIFC1 IgG binds not only to the peptide antigen but also to the intracellular full-size KIFC1, co-localization of cytosolic anti-KIFC1 IgG with GFP-KIFC1 expressed in the HeLa was observed by confocal microscopy (Figure 6A). In interphase, KIFC1 is in the nucleus and cannot interact with IgG expressed in the cytosol. KIFC1 binds to the microtubule that forms a mitotic spindle in the metaphase of mitosis and moves toward the centrosome (minus-directed). Therefore, anti-KIFC1 IgG expressed in the cytosol can bind to anti-KIFC1 in mitosis. Similar to the cell lysate ELISA results, of the three anti-KIFC1 IgGs, 6C407 and 2C281 co-localized with KIFC1 in the mitotic phase, whereas 10C358 did not (Figure 6A). In the case of cytosolic 3D8 IgG, there is no antigen that can be used in cells. To confirm whether the antigen-binding site of cytosolic 3D8 IgG was properly formed, O2F3 IgM, an anti-idiotypic antibody that recognizes the conformation of the variable region of 3D8 IgG, were used to confocal microscopy (Figure 6B). HEK293T cells expressing cytosolic 3D8 IgG were fixed and permeabilized, and then treated with O2F3 IgM and its specific fluorescent-labeled antibody. Confocal microscopy confirmed that cytosolic 3D8 IgG was recognized by O2F3, indicating
30
that the antigen binding site of cytosolic 3D8 IgG is properly formed. The results of the antigen-binding activity of cytosolic IgG showed that the variable region of the antibody determines the antigen-binding activity of cytosolic IgG.
31
Figure 5. Evaluation of binding activity of cytosolic IgGs. (A) The antigen-binding activity of cytosolic anti-KIFC1 IgG was analyzed by ELISA using peptide antigens synthesizing the epitope amino acid sequence of each antibody (KIFC1 peptide #1 for 2C281, #2 for 6C407, and #3 for 10C358). Lysates of transfectants were placed in wells coated with specific antigens and bound IgGs were detected with AP-conjugated anti-human IgG/Fc. Purified IgGs from HEK293F cells were used as positive control. (B) Binding activity of 3D8 anti-DNA IgG was analyzed by ELISA using synthetic single-strand DNA antigen. The subsequent ELISA procedure was performed in the same manner as in (A). Data are presented as mean ± SEM, n = 3. (C) An illustration showing the progress of the experiment.
32
Figure 6. Confocal microscopic analysis to confirm antigen-binding site of the cytosolic IgGs. (A) Cellular antigen-binding activity of cytosolic anti-KIFC1 IgGs were analyzed by confocal microscopy. HeLa cells stably expressing GFP-KIFC1 were transfected with the plasmids encoding ΔLd-anti-KIFC1 IgG gene. After synchronization of cells to mitotic phase, cells were fixed and stained with a primary antibody for anti-human IgG/Fc, followed by rhodamine-conjugated anti-goat IgG. (B) Formation of antigen-binding site of cytosolic 3D8 IgG was analyzed by confocal microscopy. Transiently transfected HEK293T cells were fixed, permeabilized, and then incubated with O2F3 (mouse IgM), followed by an Alexa Fluor 647-conjugated anti-mouse IgM/μ chain antibody. Bar = 10 μm.
33
C. Association of heavy and light chains of cytosolic IgGs
To explore why the antigen-binding activities of cytosolic IgGs are different each other, association between heavy and light chains of IgG was examined. We examined the association between heavy and light chains that compose the antigen binding site. Heavy chains of IgG in the HEK293T transfectants were immuno-precipitated by Protein-A/G and light chains interacting with the heavy chains were detected by western blot (Figure 7). As a result, H:L association was not observed in cytosolic 10C358 IgG that lost antigen-binding activity when expressed in the cytosol. The association of heavy and light chains of IgG was also confirmed through ELISA (Figure 8). Heavy chain of cytosolic IgG in the cell lysate is captured by Fc-specific antibody that is coated on the ELISA plate. The light chain associated with the heavy chain of cytosolic IgG is detected as a Cκ-specific antibody, and the AP-conjugated secondary antibody and the substrate are treated sequentially. ELISA was also performed to reverse the order of the antibodies used for capture and detection. After coating the Ck-specific antibody on the plate, the light chain of the cytosolic IgG was captured, and the heavy chain associated with the light chain was detected as AP-conjugated Fc-specific antibody. As a result, association between heavy and light chains was not observed in cytosolic 10C358 IgG as in IP results. These results confirm that the difference in antigen-binding of cytosolic IgG is related to the association of heavy and light chains.
34
Figure 7. Association of heavy and light chains of cytosolic IgGs. HEK293T cells lysates of IgGs transfectants were immunoprecipitated using Protein A/G-agarose. Input and IP samples were resolved by reducing SDS-PAGE and detected with goat anti-human IgG (Fc specific), goat anti-human IgG (kappa chain specific), and rabbit anti-cytoskeletal actin antibody, followed by HRP-conjugated rabbit goat IgG and HRP-conjugated goat anti-rabbit IgG, respectively. Input represents 10% of the total amount of lysate used in for IP.
35
Figure 8. Evaluation of H:L association of cytosolic IgGs. (A) Anti-human IgG/Fc was used as a capture antibody, followed by treatment of IgGs transfectants. Bound IgGs were detected by anti-human C and AP-conjugated secondary antibody. Asterisk (*) indicates negative control to ensure that the anti-rabbit IgG/Fc-specific antibody does not directly react with human IgG/Fc. (B) Anti-human C was used as a capture antibody. Lysates of HEK293T cells expressing IgGs were treated and detected by AP-conjugated anti-human IgG/Fc. Data are presented as mean ± SEM, n = 3.
36
D. H:L association in the absence of formation of the correct antigen-binding site If the antigen-binding activity of the variable region is not present, does it mean that the H:L association not occur? To explore this, hybrid 2C281 was constructed by replacing the Vκ site of cytosolic 2C281 IgG with a pseudo Vκ that does not bind to KIFC1, thereby eliminating the antigen-binding activity and comparing its characteristics with the original 2C281 IgG (Figure 9A). ELISA was performed to investigate antigen-binding activity and confirmed that the cytosolic hybrid 2C281 IgG did not bind to its peptide antigen (Figure 9B). However, the results of H:L assembly-ELISA and IP showed that the heavy and light chain assembly of the cytosolic hybrid 2C281 IgG was maintained (Figure 9C, D). It was confirmed that even though the antigen-binding site is not properly formed, H:L association can occur irrespective of the antigen-binding site formation.
37
Figure 9. Association of heavy and light chains without formation of the correct antigen-binding site. (A) Schematic representation of Ld-hybrid IgG1 possessing 2C281 VH and pseudo Vκ as variable regions. (B) Evaluation of antigen-binding activity by ELISA. Lysates of HEK293T cells expressing ΔLd-2C281 or ΔLd-hybrid IgG were treated in wells coated with KIFC1 #1 peptide and bound chimeric IgGs were detected with AP-conjugated anti-human IgG/Fc antibody. (C) Evaluation of association between heavy and light chains by ELISA. Lysates of HEK293T cells expressing ΔLd-2C281 or ΔLd-hybrid IgG were treated in wells coated with anti-human C, and bound IgGs were detected with AP-conjugated anti-human IgG/Fc antibody. In (B, C), data are presented as mean ± SEM, n = 6. (D) Co-IP to
38
confirm assembly of heavy and light chains. Lysates of HEK293T cells expressing ΔLd-2C281 or ΔLd-hybrid IgG were immunoprecipitated using Protein A/G-agarose. Proteins in the eluate were detected with goat anti-human IgG (Fc specific), goat anti-human IgG (Cκ specific), and rabbit anti-actin antibody, followed by HRP-conjugated rabbit anti-goat IgG and HRP-conjugated goat anti-rabbit IgG, respectively. Input and elution samples were resolved by reducing SDS-PAGE. Input represents 10% of the total amount of lysate used for IP.
39
E. Effect of structural integrity of the constant region on H:L association and formation of the correct antigen-binding site in cytosolic IgGs
If so, is the heavy and light chain linkage of the cytosolic IgG determined entirely by the variable region? To analyze the effect of constant region on H:L association, disulfide bonds were focused , which play an important role in heavy and light chain interaction. The mutant ΔLd-IgG *#H*#L, which cannot form all disulfide bond, was expressed by replacing all cysteine residues in the constant region with serine residues which have same structure with cysteine but one oxygen atom instead of sulfur (Figure 10A). Expression of mutant and wildtype cytosolic IgGs was confirmed by western blotting (Figure 10B) and the assembly of heavy and light chains and antigen-binding activity were confirmed by ELISA (Figure 10C, D). As a result, interaction of heavy chain-light chain and antigen-binding activity were not observed in all mutants. This shows that the constant region of cytosolic IgG may affects the H:L association and antigen-binding activity.
40
Figure 10. Effect of the absence of disulfide bonds in constant regions on H:L association and correct folding of antigen-binding site in cytosolic IgGs. (A) Schematic representation of Ld-IgG*#H*#L. Sharp (#) means disruption of intra-chain disulfide bonds and asterisk (*) means disruption of inter-chain disulfide bonds. (B) Heavy and light chains in lysates of HEK293T cells transiently transfected with KV10 vectors encoding wt ΔLd-IgG and mutant ΔLd-IgG*#
H*#L were detected by immunoblotting. Heavy chain was detected with goat anti-human IgG (Fc specific) and light chain was detected by goat anti-anti-human IgG (Cκ specific),
41
followed by HRP-conjugated rabbit anti-goat IgG. (C) ELISA to evaluate association of heavy and light chains was performed using anti-human C as a capture antibody. Lysates of HEK293T cells expressing wt ΔLd-IgG and mutant ΔLd-IgG*#H*#L were treated and bound IgGs were detected with AP-conjugated anti-human IgG/Fc antibody. (D) ELISA to evaluate antigen-binding of wt ΔLd-IgG and mutant ΔLd-IgG*#H*#L was performed. Lysates of HEK293T cells expressing wt ΔLd-IgG and mutant ΔLd-IgG*#H*#L were treated to wells coated with their specific antigens and bound IgGs were detected with AP-conjugated anti-human IgG/Fc antibody.
42
F. Importance of intra-chain disulfide bonds in H:L association and antigen-binding site formation of cytosolic IgGs
In order to understand disulfide bonds present in the constant region, antibodies were expressed without the variable region and designated as Ig-Cw (Immunoglobulin whole constant region). I constructed a *#CH*#CL mutant that does not form all disulfide bonds and *CH*CL mutant that does not form inter-chain disulfide bonds (Figure 11A). Protein expression was confirmed by immunoblotting (Figure 11B), and expression levels were extremely low when all disulfide bonds were eliminated regardless of the leader sequence. It is presumed that the absence of disulfide bonds has some influence on the stability of the protein. ELISA showed that H:L association was observed in CHCL and *CH*CL, which have all disulfide bonds and intra-chain disulfide bonds, respectively. However, heavy and light chains of *#CH*# CL, which eliminated all disulfide bonds, was not assembled (Figure 11C). These results indicated that intra-chain disulfide bonds are important to association of heavy and light chain.
43
Figure 11. Effect of intrachain disulfide bonds on H:L association and antigen-binding site formation of cytosolic IgGs. (A) Schematic representation of Ld-Ig-Cw (whole constant region) mutants. Sharp (#) means disruption of intra-chain disulfide bonds and asterisk (*) means disruption of inter-chain disulfide bonds. The black square at the N-terminus of Cw means leader sequence. (B) Heavy and light chains in lysates of HEK293T cells transiently transfected with KV10 vectors encoding Ig-Cw with or without Ld were detected by immunoblotting. Heavy chain was detected with goat anti-human IgG (Fc specific) and light chain was detected by goat anti-human IgG (C specific), followed by HRP-conjugated rabbit anti-goat IgG. (C) ELISA to evaluate association of heavy and light chains was performed using anti-human C as a capture antibody. Lysates of HEK293T cells expressing Ld-Ig-Cw and ΔLd-Ig-Cw constructs were treated and bound Ig-Cw were detected with AP-conjugated anti-human IgG/Fc antibody. Data are presented as mean ± SEM, n = 3.
44
G. Partial formation of intra-chain disulfide bonds in heavy and light chains of cytosolic IgGs
Cysteine residues in the state of reduced cytosol environment can form disulfide bonds without enzymatic reaction when cells are destroyed and exposed to an oxidizing environment in the air. To exclude this possibility, I compared the differences between the cells after exposure to air for 0 and 12 h by western blotting. The HA or Flag tag were labeled to the carboxy terminal of the heavy or light chain of 3D8 IgG and expressed in the cytosol, and the expression level was confirmed by western blotting (Figure 12A, B). Analysis showed no difference between the 0 and 12 h-exposed samples in the air. When treated with the thiol alkylating agent NEM, some cysteine residues in the IgG were alkylated and the band size was slightly increased (Figure 12B, lane 3,4). The reason for the alkylation of cytosolic IgG by NEM is that some cysteine residues exist in reduced form without forming intra-disulfide bonds. Thus, this suggests that the disulfide bond in the cytosolic IgG is partially formed. When the antigen-binding capacity of the antibody treated with NEM was determined by ELISA (Figure 12C), in the case of ER-directed IgG, most cysteine residues were already binary and there was little free-thiol. However, cytosolic IgG has a disulfide bond formed only in some cysteine residues, resulting in a decrease in antigen-binding activity due to the presence of alkylated cysteine by NEM. Therefore, it was confirmed that the cysteine residues in the cytosolic IgG partially formed disulfide bonds.
The presence of intra-disulfide bond in cytosolic IgG was confirmed by PEGylation of 3D8 IgG tagged with HA at the carboxy terminus of the heavy chain expressed in the cytosol of HEK293T and treated with NEM to block the free thiol. Then, TCEP was treated to reduce disulfide bonds and free-thiol, newly exposed by TCEP, is detected by PEGylation using
45
mPEG-MAL followed by detection of IgG heavy chain by western blot. In other words, if there is a disulfide bond in the cytosolic IgG, it can be confirmed by increased molecular weight by western blotting by labeling with PEG (Figure 13A). The NEM present in the cell lysate is removed by a desalting column prior to TCEP treatment because NEM competes with the maleimide group of mPEG-MAL. Western blot results showed that PEGylated IgG heavy chain band, slightly larger than native IgG heavy chain, was observed in lane 1 (Figure 13B). This indicates that disulfide bonds are present in cytosolic IgG.
46
Figure 12.Absence of spontaneous ex vivo non-enzymatic disulfide bonds formation in cytosolic IgGs. (A) Schematic representation of Ld-3D8 IgG/H-HA and Ld-3D8 IgG/L-Flag proteins. (B) Immunoblotting was performed using lysates of HEK293T cells transiently transfected with KV10 vectors encoding Ld-3D8 IgG/H-HA or Ld-3D8 IgG/L-Flag and prepared in the presence and absence of 100 mM NEM. Proteins were separated by non-reducing SDS-PAGE, both immediately and after 12 h incubation at RT. HA-tagged heavy chain was detected with mouse anti-HA tag antibody and Flag-tagged light chain was detected by moues anti-Flag tag antibody, followed by HRP-conjugated horse anti-mouse IgG. (C) antigen-binding activity of IgGs was evaluated by ELISA. Lysates of HEK293T cells transiently transfected with Ld- or ΔLd-IgGs were prepared in the presence of 100 mM NEM, placed in wells coated with specific antigens. Bound IgGs were detected with AP-conjugated anti-human IgG/Fc. Data are presented as mean ± SEM (n = 6).
47
Figure 13. Presence of disulfide bonds in the cytosolic IgG. (A) Workflow of the assay. (B) Immunoblotting. HEK293T cells transfected with a plasmid encoding Ld-3D8 IgG/H-HA were pre-incubated with NEM at a final concentration of 50 mM at 37C for 15 min. Lysates of cells were then prepared in the presence of 100 mM NEM. NEM was removed by passing the cell lysate through a desalting column to avoid competition with mPEG-MAL. Lysates were reacted with 5 mM TCEP at 37°C for 1 h and then incubated with 10 mM mPEG-MAL at 37°C for 1 h. PEGylated proteins were separated by reducing SDS-PAGE, and HA labeled heavy chain and PEG were detected by immunoblotting with anti-HA and anti-PEG antibodies, respectively. The PEGylated 3D8 IgG heavy chain is indicated by an arrow.
48
H. Differences in assembly of IgG1s expressed in the cytosol or via the ER
To determine the difference in disulfide bond formation between cytosolic IgG and ER-directed proteins, in vitro reversible redox reactions were performed. The IgG-expressing HEK293T transfectants were treated with 100 mM of DTT to completely reduce the IgG, and the solution was passed through a desalting column to remove DTT. Following this, micro-dialysis was performed to make the protein fold, and change in the IgG was confirmed by immunoblotting. It is confirmed that DTT could be removed by passing desalting column and dialyzing (Figure 14A). In the DTT-treated sample, all disulfide bonds of IgG were reduced, and a single band was observed regardless of the leader sequence (Figure 14B, lane 3). Upon removal of DTT, heavy chain of both ΔLd-3D8 IgG and Ld-3D8 IgG returned to the state as before DTT treatment (Figure 14B, lane 2). Western blot analysis of Ld-3D8 IgG revealed that several bands were observed at higher than 50 kDa, indicating that inter-chain disulfide bonds were present. In the case of ΔLd-3D8 IgG, only a 50 kDa size band was observed after DTT removal, so it is considered that only intra-chain disulfide bonds are formed. Although ΔLd-3D8 IgG is spontaneously oxidized in the air for 24 h, it did not form inter-chain disulfide bonds, unlike Ld-3D8 IgG. This suggests that although Ld-3D8 IgG and ΔLd-3D8 IgG have the same amino acid sequence, they have different protein conformations depending on the initial expression environment.
49
Figure 14. Comparison of assembly patterns of IgG expressed in reduced cytosol and through oxidative environment of ER. (A) The concentration of free thiols in PBS was measured before and after DTT removal steps. (B) In vitro reversible redox reaction was performed to analyze the association pattern of heavy and light chains following DTT removal. Lysates of HEK293T cells transfected with KV10ΔLd-3D8 IgG or KV10Ld-3D8 IgG were treated with 100 mM DTT for 30 min at RT, followed by DTT removal steps comprising passage through a desalting column and dialysis against PBS at 4°C for 24 h. Lysates were separated by non-reducing SDS-PAGE, and the heavy chain was detected by immunoblotting with anti-IgG/Fc antibody.
50
I. Antigen-binding affinity of purified chimeric anti-KIFC1 IgGs
To measure the antigen-binding affinity of anti-KIFC1 IgGs expressed in the ER and secreted out of the cell, chimeric anti-KIFC1 IgGs were expressed and purified in mammalian cells. Chimeric anti-KIFC1 IgGs were expressed in HEK293F cells and culture supernatants were subjected to Protein-A affinity chromatography to purify IgG proteins. 2C281 and 6C407 IgG, which exhibited antigen-binding activity when expressed in the cytosol, were purified and their purity and size were confirmed by SDS-PAGE (Figure 15). The antigen-binding affinity of the anti-KIFC1 chimeric IgGs were measured using octet. Biotinylated peptides used as antigen were composed of from 1 to 50 amino acid of KIFC1 for 6C407 IgG or from 51 to 100 amino acid of KIFC1 for 2C281 IgG. Immobilized peptide antigens on the sensor were reacted with purified anti-KIFC1 antibodies. As a result, antigen-binding affinity of 2C281 IgG and 6C407 IgG were showed about three times higher binding capacity than 6C407 (Figure 16).
The scFv form of antibodies is widely seen in intracellular antibodies because of its concise molecular size. Prior to analysis of cytosolic anti-KIFC1 scFvs, I confirmed antigen-binding activity of antibodies composed of variable regions in heavy and light chains. Antibodies in the form of scFv were constructed by linking the variable region of the heavy and light chains by 15 amino acids (Gly4Ser)3 linker (Figure 17A). The anti-KIFC1 antibody containing the Protein-A tag was expressed in the periplasmic space of Escherichia coli and purified by IgG-sepharose affinity chromatography, and its approximate molecular weight was confirmed by SDS-PAGE (Figure 17B). In the presence of DTT, scFv proteins were located slightly higher than DTT-untreated proteins, indicating possible intra-chain disulfide bonds.
51
The binding affinity of the two anti-KIFC1 scFv proteins to the antigen was measured quantitatively by octet. Streptavidin sensor was coated with biotinylated KIFC1 peptide and reacted with various concentrations of purified scFv proteins and the binding of scFvs to peptide-coated sensor was measured (Figure 17C). The dissociation level was observed by reacting with a solution containing no scFv, and KD value was calculated (Table 3). As a result, both antibodies had high antigen-binding affinity and the antigen-binding affinity of 2C281 was about 1.6 times higher than that of 6C407.
52
Figure 15. Purification of chimeric anti-KIFC1 IgGs. (A) Schematic diagram of chimeric anti-KIFC1 IgG. Variable region is derived from mouse hybridoma antibody and constant region is derived from human IgG1. (B) Purified chimeric anti-KIFC1 IgGs (3 μg) were separated by SDS-PAGE gel in reducing or non-reducing condition. IgGs expressed in the culture supernatant of HEK293F cells were purified using protein-A affinity column. In the non-reducing condition, the size of the full-length IgG was about 150 kDa, and under reducing condition, the heavy chain of 50 kDa and the light chain of 25 kDa were identified by SDS-PAGE and then stained with Coomassie blue.
53
Figure 16. Measurement of antigen-binding affinity of purified chimeric anti-KIFC1 IgGs. Antigen-binding affinity of anti-KIFC1 IgGs were measured by octet systems. Purified 2C281 (A) and 6C407 (B) IgGs that were serially diluted 2-fold starting at 100 and 50 nM concentration were analyzed. Biotinylated KIFC1 peptide antigens were immobilized on a streptavidin sensor, and the association rates of antibodies were measured by dipping the sensor in a well containing diluted proteins. The dissociation rate was then measured by immersing the sensor in a buffer without protein. The sensorgram, association rates (kon), dissociation rates (koff), and dissociation constant (KD) were shown.