INTRODUCTION
DNA microarrays have found widespread use as a flexi-ble tool to investigate bacterial metabolism. DNA micro-arrays provide ‘global views’ of biological processes, which facilitate comprehensive description of the metabolic and physiological state of an entire organism under a given condi-tion or genetically manipulated background. This allows microbiologists to monitor metabolism and to define stimu-lons and regustimu-lons. They offer a more holistic approach to study cellular physiology and therefore complement the traditional “gene-by-gene” approaches. The importance of this technology increases with the number of sequenced genomes. Therefore, analysis of the transcriptome using
DNA microarrays has become a standard approach for investigation of the molecular basis of microbial genetics (Ehrenreich 2006).
Deinococcus radiodurans is a red-pigmented, nonpatho-genic, nonsporulating, obligate aerobic gram-positive bac-terium. It is extremely resistant to an ionizing radiation, UV light, hydrogen peroxide, and numerous other agents that damage DNA. The complete genome sequence of D. radio-durans R1 was determined in 1999. The genome is com-posed of two chromosomes (2,648,638 and 412,348 bp), a megaplasmid (177,466 bp), and a small plasmid (45,704 bp), yielding a total genome of 3,284, 156 bp. The genome contains 3,187 ORFs, with an average size of 937 bp, repre-senting 91% of the genome. However, analysis of the geno-me sequence has not elucidated the comprehensive genetic basis of the DNA repair capabilities (White et al. 1999; Makarova et al. 2001; Edwards and Battista 2003).
─ ─ 53 ──
DNA Microarray Fabrication of Deinococcus radiodurans
R1 for a Global Gene Expression Profiling
Min-Ho Joe, Sang-Young Lim, Sun-Wook Jung, Young-Ji Choi, Bo-Young Lee, Du-Sub Song, Jin-Woo Jung, Sung-Hwa Chae1and Dong-Ho Kim*
Radiation Research Center for Biotechnology,
Korea Atomic Energy Research Institute, Jeongeup 580-185, Korea 1Research Institute of GnC BIO Co. Ltd., Daejeon 305-801, Korea
Abstract -- DNA microarray technology is an extraordinarily powerful technique, which has revolutionized the world of science and technology more profoundly than any other recent major advance. In this study, we fabricated a DNA microarray chip of Deinococcus radiodurans R1 to get the comprehensive information of this organism responding to diverse conditions. Among 3,187 open reading frames (ORFs), the PCR products representing 3,116 ORFs were obtained by using polymerase chain reactions (PCRs) with gene-specific primer sets. The PCR products were spotted in duplicate onto a slide and immobilized successfully. Hybridization experiment showed an enough sensitivity and reliability of the DNA chip to analyze global gene expression of D.
radiodurans R1. Finally, a clone library of the PCR products was constructed for convenience in
later use. The success rate of the library construction was about 95%, meaning that 3,031 ORFs were cloned into a vector.
Key words : DNA microarray, Deinococcus radiodurans R1, Global gene expression
* Corresponding author: Dong-Ho Kim, Tel. +82-63-570-3140, Fax. +82-63-570-3149, E-mail. [email protected]
Recently, several researchers have documented the chan-ges in genome-wide gene expression of D. radiodurans (Liu
et al. 2003; Tanaka et al. 2004; Schmid et al. 2005; Wang et al. 2008; Chen et al. 2008). The first global
transcrip-tomic analysis of D. radiodurans was done by Liu et al. (2003) on the time course repertoire of genes responsible for an acute irradiation after a dose of 15 kGy. Secondly, Tanaka et al. (2004) demonstrated that the global transcrip-tional responses to radiation and desiccation are correlated with each other. In addition, they discussed the recA-depen-dent and -indepenrecA-depen-dent recovery processes of D.
radiodu-rans after a sublethal dose of ionizing radiation. Other
studies were focused on the defining of regulons of specific transcriptional regulators such as an extracytoplasmic function-type heat shock sigma factor (Sig1), a novel radia-tion response regulator (DrRRA), and a sensory transcrip-tional regulator (OxyR) (Schmid et al. 2005; Wang et al. 2008; Chen et al. 2008). Taken together, transcriptomic analysis of D. radiodurans could make it possible to reveal the complex recovery process after ionizing radiation which is unique to D. radiodurans and to identify numerous genes that underlie the exceptional radiation resistance of this bacterium.
In this study, we fabricated a DNA microarray chip and a PCR product clone library for genome-wide gene expres-sion analyses of D. radiodurans R1 in various environ-ments.
MATERIALS AND METHODS
Bacterial strains, media, chemicals, and growth conditions
D. radiodurans R1 ATCC13939 was grown at 30�C in TGY (0.5% tryptone, 0.3% yeast extract, 0.1% glucose) broth or on TGY agar (1.5% agar) (Mattimore et al. 1995; Earl et al. 2002). In liquid culture, cell density was determined at 600 nm by a Beckman Coulter spectrophotometer. Escherichia coli strains were grown at 37�C in Luria-Bertani broth or on agar. Ampicillin (100μg ml-1), isopropyl- β-D-1-thio-galactoside (0.5 mM), and 5-bromo-4-chloro-3-indolyl- β-D-galactopyranoside (80μg ml-1) was added to culture media as appropriate. Transformations of E. coli were performed using commercially available cells (TOP10 [Invitrogen] and JM109 [Promega]). Chemicals were
purchased from Sigma and Aldrich chemical company.
Chromosomal DNA extraction
TGY broth (200 ml) was inoculated with a 2 ml of over-night culture (2×107CFU ml-1) of D. radiodurans R1. After 48 h, 200 ml of the cultures were harvested by centri-fugation at 4�C at 4,000 rpm for 10 min. Pellets were resus-pended in 20 ml of 95% ethanol and held at room tempera-ture for 10 min to remove the outer membrane of D. radio-durans. The ethanol-stripped cells were collected by centri-fugation at 4�C at 4,000 rpm for 10 min, and the resulting pellet was resuspended in 9 ml of TE buffer (10 mM Tris-HCl, 0.1 mM EDTA; pH 8.0). Two milligrams of lysozyme was added to stripped cells, and this mixture was incubated at 37�C for 30 min. A 0.5 ml volume of 10% sodium dode-cyl sulfate and 50μl of proteinase K (20 mg ml-1) were added to lysozyme-treated cells, and the mixture was incubated for 12 h at 56�C. Lysed cells were transferred to a centrifugal tube and extracted once with an equal volume of phenol-chloroform (1 : 1) and twice with equal volumes of chloroform-isoamyl alcohol (24 : 1). The DNA was preci-pitated by adding 1 ml of 3 M sodium acetate (pH 7.0) and 20 ml of ice-cold 100% ethanol to the extracted material. The DNA was spooled out with a curved glass rod and washed twice with 70% ethanol. The DNA was air dried, dissolved in 5 ml of TE buffer (pH 8.0), and stored at -20�C.
Total RNA extraction
Total RNA was extracted from 25 ml cultures of D. radio-durans R1 at lag, early exponential, and late exponential phase. Cultures were harvested by centrifugation at 4�C at 4,000 rpm for 10 min. Pellets were resuspended in RiboEx reagent (Geneall Biotechnology), mechanically disrupted with glass beads, and the total RNA samples were extracted by following the manufacturer’s instructions. Isolated total RNA samples were further purified with an RNeasy Mini-kit and an RNase-Free DNase I set (QIAGEN). RNA quality and quantity were evaluated using a 2,100 Bioanalyzer (Agilent Biotechnologies) by following the supplier’s pro-tocol.
Microarray design and construction
in the fully sequenced R1 genome (White et al. 1999). PCR products represent internal portions of annotated sequences with a size range between 200 and 300 bp. Primer pairs were designed for 3,187 ORFs by Illumina. PCR products were generated by combining 20 ng of genomic DNA from strain R1 with oligonucleotide primer pairs (20 pmol each, average Tm==58�C) and AccuPower� HF PCR PreMix (Bioneer) in a total volume of 50μl. The other reaction components were as specified by the manufacturer. PCR amplification successes were scored (single band and cor-rect size). Failed reactions were repeated 3 times at dif-ferent reaction conditions or with new primer pairs. The amplified PCR products (50μl) were purified using a liquid handler (TECAN) and Life Science AcroPrepTM96 Filter Plate Omega 10 K plate column (Pall). The purified PCR products were visualized by gel electrophoresis in the pre-sence of ethidium bromide, and their respective sizes were verified by comparison to the expected product length. PCR products were spotted onto Corning GAPS II (Corn-ing Life Sciences) us(Corn-ing a Gene Machine (Genomic Solu-tion Inc.). PCR products were immobilized to the slide surface using a Stratalinker UV crosslinker (Stratagene). All slides were stored in a desiccator at room temperature.
Probe preparation, microarray hybridization, and data analysis
cDNA probes for a microarray hybridization were pre-pared from three biological replicate total RNA samples. Ten micrograms of total RNA from each growth phase was annealed to 9μg of random hexamer primers (Invitrogen) in a total volume of 31μl by incubating it for 10 min at 70�C and subsequently keeping it on ice for 2 min. cDNA was synthesized at 42�C for 2 h in 50μl of the reaction mixtures containing 10μl of 5× first-strand buffer, 10 mM dithio-threitol, 0.5 mM dNTP mix containing amino allyl-dUTP (Amersham Biosciences), and 500 units of SuperScript II reverse transcriptase (Invitrogen). The reaction was termi-nated by adding 16.5μl EDTA (0.5 M, pH 8.0) and the RNA was hydrolysed by adding 16.5μl of NaOH (1 M), incuba-ting at 65�C for 15 min, and then neutralizing with 16.5μl of HCl (1 M). Unincorporated free amino allyl-dUTPs were removed by ultrafiltration with YM 30 (Millipore), and the resultant cDNA samples were coupled to 1 pmol Cy3 or Cy5 dyes (Amersham Biosciences) in 0.1 M sodium
car-bonate buffer (pH 9.0) for 2 h at room temperature in the dark. Unincorporated dyes were removed by passage over QIAquick MinElute PCR purification columns (QIAGEN). Labeled cDNA samples were mixed, dried using rotary speed vacuum, dissolved with 50μl of hybridization buffer containing 50% formamide, 5× SSC, 0.1% SDS, 5μg of hu-man Cot-1 DNA, and 5× denhardt’s solution. Labeled pro-bes were denatured by heating at 95�C for 5 min and hybri-dized to a prehybrihybri-dized DNA microarray slide (1 h incuba-tion at 42�C in 25% formamide, 5× SSC, 0.1% SDS, and 1% bovine serum albumin) for 16 h at 42�C. After hybri-dization, the slide was washed sequentially for 10 min each in 2× SSC/0.1% SDS buffer, 0.1× SSC/0.1% SDS buffer, and 0.1× SSC buffer, rinsed in distilled water for 2 min, and dried by centrifugation for 5 min at 700 rpm in a 50 ml uncapped centrifugal tube.
The hybridized slide was scanned on a GenePix 4000B imager (Axon) at both 532 nm and 635 nm visible light. Spot intensities and background signals were quantified for each channel using GenePix pro 6. Normalization and statistical analysis were carried out with Genespring soft-ware package (Agilent).
Library construction of PCR products
The purified PCR products were ligated into an yT & A vector and transformed into E. coli (RBC Bioscience). Success of a cloning was confirmed by colony PCR with M13 forward (5′-CGC CAG GGT TTT CCC AGT CAC GA-3′) and reverse (5′-AGC GGA TAA CAA TTT CAC ACA GGA-3′) primers. And 384 randomly selected plas-mids from the library were isolated from E. coli cells by using the alkaline lysis method (Sambrook and Russell 2001) and sequenced in both directions with M13 forward and reverse primers using an ABI 3,730 DNA analyzer (Applied Biosystems).
RESULTS AND DISCUSSION
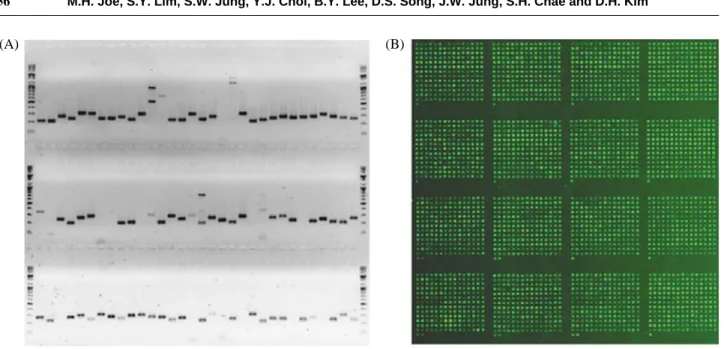
Primer design, amplification, and spottingTo construct DNA microarray, we designed a gene-speci-fic primer (18 to 22 mer) set for each 3,187 ORF. From the first round of PCRs, 75% (2,371 ORFs) of the PCR prod-ucts showing single band with correct size were obtained.
Failed reactions showing no band, multi bands, or a band with an incorrect size after agarose gel electrophoresis were repeated at different reaction conditions or with a new primer set (Fig. 1A). After 4 additional rounds of PCRs, the success rate of the PCR amplification was achieved up to 97.8% (3,116 ORFs). According to the previous reports, success rates of PCRs were from 93% to 95% (Liu et al. 2003; Tanaka et al. 2004; Schmid et al. 2005; Wang et al. 2008). Therefore, this is the best result compared to other reports.
The purified PCR products were spotted as duplicated sets of 3,116 PCR products onto amino-silane coated slides at a concentration of 100 ng μl-1. The resulting microarray chip was named WGM-DR1 (Whole genome microarray-D.
radiodurans R1). The spot quality on the WGM-DR1 chip
was confirmed by the homogeneous spot intensity obtained after Syber Green 2 staining (Fig. 1B).
Quality confirmation of the microarray chip
For a further quality confirmation, we performed a self-against-self hybridization experiment. cDNA probes were prepared from the same amount of total RNA isolated from
D. radiodurans R1 cells at three different growth phases to
cover the whole RNA molecules. The self-self hybridiza-tion should ideally produce similar signal intensity in both channels for every spot. Global normalization was applied to normalize the Cy5 signal intensities against Cy3, on an
assumption that the total intensity of the Cy5 channel is equal to that of Cy3 (Bilban et al. 2002; Yang et al. 2002). The scatter plot showed tight signal intensities in both channels (Fig. 2). Therefore, the quality of the DNA micro-array chip is sufficient for a transcriptomic analysis. How-ever, the result also indicated that the whole process, inclu-ding the target labeling and hybridization procedure, need Fig. 1. An image of a representative agarose gel separation containing 96-PCR samples (A) and Syber Green 2 staining of microarray slide
after spotting (B).
(A) (B)
Fig. 2. Scatter plot showing the distribution of Cy3 and Cy5 signal intensities from a self-self hybridization experiment.
to be optimized for a better result.
PCR clone library construction
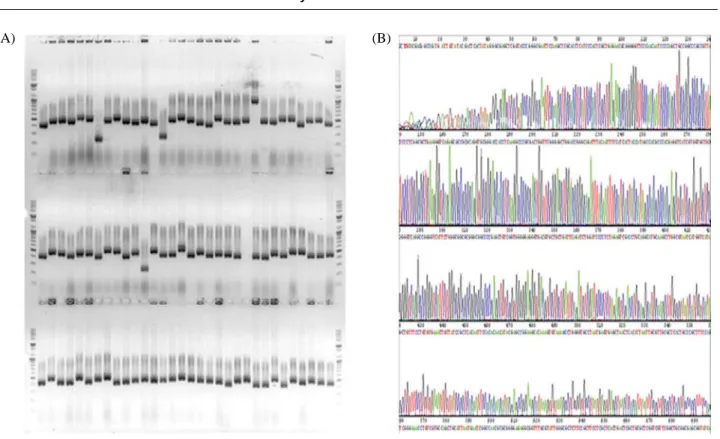
For a convenience in a later use, we constructed a clone library of the PCR products. The vector, yT & A, used in this experiment has priming sites corresponding to M13 uni-versal primers at both ends of the cloning sites. Therefore, PCR products for a DNA chip fabrication can be obtained easily. The success of cloning was confirmed by colony PCR (Fig. 3A). After three rounds of cloning reactions, a PCR clone library was constructed containing 3,031 clones. For the DNA sequence accuracy of the PCR clones, DNA sequence of 384 randomly selected clones were determined (Fig. 3B). The sequencing result showed a few negligible single nucleotide mismatches. Therefore, we finally made a PCR clone library containing 95% of the whole ORFs of D.
radiodurans R1.
ACKNOWLEDGEMENT
This study was supported by Ministry of Education, Sci-ence & Technology (MEST), Korean government, through
its National Nuclear Technology Program.
REFERENCES
Bilban M, Buehler LK, Head S, Desoye G and Quaranta V. 2002. Normalizing DNA microarray data. Curr. Issues Mol.
Biol. 4:57-64.
Chen H, Xu G, Zhao Y, Tian B, Lu H, Yu X, Xu Z, Ying N, Hu S and Hua Y. 2008. A Novel OxyR sensor and regulator of hydrogen peroxide stress with one cysteine residue in
Deinococcus radiodurans. PLoS ONE. 3:e1602.
Earl AM, Mohundro MM, Mian IS and Battista JR. 2002. The IrrE protein of Deinococcus radiodurans R1 is a novel re-gulator of recA expression. J. Bacteriol. 184:6216-6224. Edwards JS and Battista JR. 2003. Using DNA microarray data
to understand the ionizing radiation resistance of
Deino-coccus radiodurans. Trends Biotechnol. 21:381-382.
Ehrenreich A. 2006. DNA microarray technology for the micro-biologist: an overview. Appl. Microbiol. Biotechnol. 73: 255-273.
Liu YZJ, Omelchenko MV, Beliaev AS, Venkateswaran A, Stair J, Wu L, Thompson DK, Xu D, Rogozin IB, Gaida-makova EK, Zhai M, Makarova KS, Koonin EV and Daly MJ. 2003. Transcriptome dynamics of Deinococcus
radio-durans recovering from ionizing radiation. Proc. Natl.
Fig. 3. A representative image of colony PCR (A) and DNA sequencing (B) results for PCR-clone library construction.
Acad. Sci. 100:4191-4196.
Makarova KS, L. Aravind L, Wolf YI, Tatusov RL, Minton KW, Koonin EV and Daly MJ. 2001. Genome of the extre-mely radiation-resistant bacterium Deinococcus
radiodu-rans viewed from the perspective of comparative
geno-mics. Microbiol. Mol. Biol. Rev. 65:44-79.
Mattimore V, Udupa KS, Berne GA and Battista JR. 1995. Genetic characterization of forty ionizing radiation-sensi-tive strains of Deinococcus radiodurans: linkage informa-tion from transformainforma-tion. J. Bacteriol. 177:5232-5237. Sambrook J and Russell DW. 2001. Molecular cloning: a
labora-tory manual, 3rd ed. Cold Spring Harbor Laboralabora-tory Press, Cold Spring Harbor, N.Y.
Schmid AK, Howell HA, Battista JR, Peterson SN and Lid-strom ME. 2005. Global transcriptional and proteomic analysis of the Sig1 heat shock regulon of Deinococcus
radiodurans. J. Bacteriol. 187:3339-3351.
Tanaka M, Earl AM, Howell HA, Park MJ, Eisen JA, Peterson SN and Battista JR. 2004. Analysis of Deinococcus
radio-durans’s transcriptional response to ionizing radiation and
desiccation reveals novel proteins that contribute to extreme
radioresistance. Genetics. 168:21-33.
Wang L, Xu G, Chen H, Zhao Y, Xu N, Tian B and Hua Y. 2008. DrRRA: a novel response regulator essential for the extreme radioresistance of Deinococcus radiodurans. Mol.
Microbiol. 67:1211-1222.
White O, Eisen JA, Heidelberg JF, Hickey EK, Peterson JD, Robert JD, Daniel HH, Michelle LG, William CN, Del-wood LR, Kelly SM, Haiying Q, Lingxia J, Wanda P, Marie C, Mian S, Jessica JV, Peter L, Lisa M, Terry U, Celeste Z, Kira SM, Aravind L, Michael JD, Kenneth WM, Robert DF, Karen AK, Karen EN, Steven S, Hamilton OS, Craig VJ and Claire MF. 1999. Genome sequence of the radioresis-tant bacterium Deinococcus radiodurans R1. Science 286: 1571-1577.
Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngal J and Speed TP. 2002. Normalization for cDNA microarray data: A robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 30:e15.
Manuscript Received: April 11, 2008 Revision Accepted: April 28, 2008