저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Nuclear shape alteration by oxidative stress
by
Ju-Hyun Ahn
Major in Cancer Biology
Department of Biomedical Sciences
The Graduate School, Ajou University
Nuclear shape alteration by oxidative stress
by
Ju-Hyun Ahn
A Dissertation Submitted to The Graduate School of
Ajou University in Partial Fulfillment of the Requirements for the
Degree of
Ph.D of Biomedical Sciences
Supervised by
Jae-Ho Lee, M.D., Ph.D.
Major in Cancer Biology
Department of Biomedical Sciences
The Graduate School, Ajou University
This certifies that the dissertation of
Ju-Hyun Ahn is approved
SUPERVISORY COMMITTEE
The Graduate School, Ajou University
June, 22
th, 2018
-ABSTRACT-
Many types of cancer exhibit abnormal nuclear shapes and increased reactive oxygen species (ROS) level compared to normal cells. However, whether ROS induce nuclear deformation has not been fully addressed. Here, hydrogen peroxide (H2O2) treatment
induced concentration-dependent alterations in nuclear shape that were abolished by pretreatment with the antioxidant N-acetyl-L-cysteine (NAC) or overexpression of catalase. Interestingly, treatment with H2O2 during mitosis induced nuclear shape
alterations significantly more compared with treatment to asynchronous cells, suggesting that nuclear shape alteration by H2O2 was mainly due to its effect on nuclear envelope
disassembly and/or reassembly processes. Because protein phosphatase 2A (PP2A) activity is reported to be involved in nuclear envelope reassembly during mitosis, the possibility of PP2A involved in nuclear shape alteration has been investigated. Indeed, H2O2 reduced the activity of PP2A and okadaic acid, a PP1 and PP2A inhibitor, induced
nuclear shape alteration. Moreover, overexpression of PP2A, but not PP1 or PP4, partially overcame H2O2-induced alterations in nuclear shape, indicating that a decrease in PP2A
activity induced by H2O2 is specifically involved in the observed nuclear shape alterations.
I also observed that the treatment of mitotic cells with H2O2 formed lamin aggregates
during early mitosis, and it correlated with the abnormal nucleus formation. Since phosphorylation by Cdk1 is important in lamin disassembly during mitosis, I wondered whether Cdk1 was involved in the formation of lamin aggregates by H2O2. Treatment of
mitotic cells with RO3306, an inhibitor of Cdk1, also induced lamin aggregates and an abnormal nucleus. Moreover, in vitro kinase assay and immunoprecipitation study demonstrated that H2O2 reduced the activity of Cdk1 via decreasing its interaction with
cyclin B. In addition, the reduction of Cdk1 activity induced prematurely lamin reassembly, which seems to contribute to the formation of abnormal nuclei.
Collectively, during early mitosis, inhibition of the Cdk1 activity by H2O2 leads to lamin
aggregates seem to affect the lamin reassembly process and eventually contribute to the formation of abnormal nucleus. In addition, during mitotic exit, H2O2 inhibits the activity
of PP2A, which plays an important role in nuclear envelope reassembly, resulting in the formation of the abnormal nucleus. Therefore, I propose that the inhibition of Cdk1 and PP2A activity by H2O2 during mitotic entry and exit, respectively, is the underlying
mechanisms involved in the change of nuclear shape.
TABLE OF CONTENTS
ABSTRACT. . . i
TABLE OF CONTENTS. . . iii
LIST OF FIGURES . . . vi
LIST OF TABLES . . . viii
ABBREVIATIONS . . . ix
I. INTRODUCTION . . . 1
II. MATERIALS AND METHODS . . . 16
A. Antibodies . . . 16
B. Cell culture . . . 16
C. Synchronization and drug treatment . . . 17
D. H2O2 treatment . . . 17
E. Determination of intracellular ROS level . . . 17
F. Measurements of intracellular level of H2O2 in living cells . . . 18
G. Plasmids and transfection experiment . . . 18
H. Transmission electron microscopy . . . 19
I. Immunocytochemistry (ICC). . . 19
J. Immunoblotting . . . 19
L. Time-lapse microscopic analysis . . . 20
M. In vitro Cdk1 kinase assay . . . 21
N. Immunoprecipitation (IP) . . . 21
O. Phos-tag analysis . . . 22
P. S-glutathionylation assay . . . 22
Q. Statistical analysis . . . 23
III. RESULTS PART Ⅰ. Inhibition of PP2A activity by H2O2 during mitosis disrupts nuclear envelope reassembly and alters nuclear shape. A. Treatment of mitotic cells with H2O2 induces abnormal nucleation . . . 24
B. Formation of abnormal nuclei following H2O2 treatment is prevented by NAC or catalase . . . 33
C. Quantitative changes in lamin B1, structural changes in the cytoskeleton and ER, and DNA damage are not major contributors to H2O2-induced nuclear shape changes . . . 36
D. H2O2 inhibits PP2A activity during mitosis . . . 41
E. Ectopic expression of PP2A rescues H2O2-induced aberrant nuclear shape changes . . . 44
F. Inhibition of PP2A activity by H2O2 causes mislocalization of core proteins during mitotic exit . . . 47
PART Ⅱ. Oxidative stress during early mitosis induces formation of aggregated lamin through decreasing Cdk1 activity, leading to abnormal nuclear shape.
A. Lamin aggregates are formed by H2O2 treatment during mitosis . . . 53
B. The lamin aggregates formed during mitosis correlates to nuclear shape changes . . . 58
C. The activity of Cdk1 is reduced by H2O2 treatment during early mitosis . . . 62
D. The inhibition of Cdk1 by H2O2 during early mitosis reduces the phosphorylation of the substrates, especially lamin protein . . . 65
E. Ectopic expression of Cdk1 rescues H2O2-induced lamin aggregates formation and aberrant nuclear shape changes . . . 70
F. Cdc25 is not involved in the inhibition of the activity of Cdk1 by H2O2 . . . . 73
G. Glutathionylation is not related to inhibition of Cdk1 activity by H2O2 . . . . 76
H. Analysis of cysteine modification of Cdk1 119 residue using mass spectrometry . . . 79
I. Reduction in the activity of Cdk1 by H2O2 during mitosis leads to nuclear envelope reassembly earlier than normal . . . 81
J. Progerin expression induces nuclear shape alteration in interphase cells and lamin aggregates in mitotic cells . . . . . . 84
IV. DISCUSSION . . . 87
REFERENCES . . . 100
LIST OF FIGURES
Fig. 1. The structure of the nuclear envelope . . . 3
Fig. 2. Reactive oxygen species and oxidative post-translational modifications (Ox-PTMs) on cysteine . . . 6
Fig. 3. Cell cycle regulation and Cdk1 activation . . . 8
Fig. 4. NE disassembly and reassembly . . . 11
Fig. 5. Protein phosphatases in nuclear envelope reassembly . . . 13
Fig. 6. H2O2 treatment to mitotic cells forms more abnormal nuclear shape than asynchronous cells . . . 28
Fig. 7. Nuclear shape alteration induced by H2O2 treatment during mitosis is rescued by antioxidants . . . 35
Fig. 8. H2O2 - induced abnormal nuclear shape is mainly neither due to changes in lamin B1 level, nor in cytoskeleton network, nor in DNA damage . . . 40
Fig. 9. Inhibition of protein phosphatase 2A activity is involved in H2O2 - induced abnormal nuclear formation . . . 43
Fig. 10. PP2A, but not PP1, overexpression overcome the H2O2 - induced nuclear shape alteration . . . 46
Fig. 11. Inhibition of PP2A by H2O2 influences BAF dynamics and nuclear envelope formation . . . 51
Fig. 12. H2O2 treatment in early mitosis induces formation of lamin aggregates . . . 57
Fig. 13. Formation of H2O2-induced lamin aggregates during mitosis seems to induce an abnormal nucleus in interphase cells . . . 61
Fig. 14. Cdk1 activity is decreased by H2O2 during early mitosis . . . 64
Fig. 15. Cdk1 inhibition by H2O2 during early mitosis prevents cells from reaching the
substrate phosphorylation threshold, in particular that of lamin . . . .. . . 69 Fig. 16. Overexpression of Cdk1 rescues formation of lamin aggregates and abnormal nuclear shape induced by H2O2 treatment . . . 72
Fig. 17. Cdc25 is not involved in inhibition of Cdk1 by H2O2 during early mitosis . . . . 75
Fig. 18. Glutathionylation is not involved in decrease of Cdk1 activity by H2O2 . . . 78
Fig. 19. Mass spectrometry analysis of Cdk1 cysteine 119th residue . . . 80 Fig. 20. The inhibition of Cdk1 activity by H2O2 accelerates the reassembly process of
lamin B1 . . . 83 Fig. 21. Cells expressing progerin have aggregates during mitosis, possibly resulting in abnormal nucleation . . . 86 Fig. 22. Schematic diagram showing that inhibition of PP2A activity during mitosis by ROS results in abnormal nuclear shape via mislocalization of BAF, a substrate of PP2A during mitotic exit . . . 89 Fig. 23. Schematic diagram showing that inhibition of Cdk1 activity during mitosis by H2O2 induces formation of aggregated lamin, leading to abnormal nuclear
shape . . . .. . . 92 Fig. 24. Cdk1 and PP2A contribute to the formation of the lamin aggregates by counterbalancing during mitosis . . . 95 Fig. 25. Schematic diagram showing how H2O2 induces abnormal nuclear shape via both
LIST OF TABLES
Table 1. Localization of lamin A/C by BAF localization in the telophase chromosome core region . . . 52
ABBREVIATION
ROS : Reactive oxygen species H2O2 : Hydrogen peroxide
NAC : N-acetyl-L-cysteine PP2A : Protein phosphatase 2A BAF : barrier-to-autointegration factor
NEBD : Nuclear envelope break down Cdk1 : Cyclin-dependent kinase 1
I. INTRODUCTION
A. Nuclear envelope (NE) and lamins
The nuclear envelope (NE) is a double membrane that surrounds the chromosomes and contains embedded nuclear pore complexes (NPCs). The outer nuclear membrane (ONM) and the inner nuclear membrane (INM) are separated by the perinuclear space and each has its own complement of inserted proteins (Fig. 1) (Hetzer et al., 2005; Stewart et al., 2007; Guttinger et al., 2009).
Some proteins in the INM interact with chromosomes or lamin proteins, thus connecting the nuclear envelope to the chromosomes or to the nuclear lamina, respectively. For example, the lamin B receptor (LBR) interacts with B-type lamins and chromatin-associated heterochromatin protein 1 (HP1) (Pyrpasopoulou et al., 1996; Polioudaki et al., 2001). LEM (lamina-associated protein 2 (LAP2), Emerin, MAN1)-domain family binds to lamin and also interacts with chromatin through a barrier-to-autointegration factor (BAF) (Margalit et al., 2007). In addition, SUN protein present in INM binds to nesprin in ONM to form LINC complex, which connects cytoplasmic cytoskeleton and nuclear lamina (Crisp et al., 2006; Walters et al., 2012).
The ONM is continuous with the endoplasmic reticulum (ER), and lumen between the ONM and INM is continuous with the ER lumen. The ONM contains several proteins that connect the nucleus to cytoplasmic filament systems and the centriole, potentially contributing to cell polarity and mobility (Dahl et al., 2008; Lombardi et al., 2011).
Lamins are generally divided into two types, A-type and B-type. In mammals, three genes,
LMNA, LMNB1, and LMNB2, encode lamin proteins. LMNB1 encodes lamin B1 protein, which
is expressed in most somatic cells. LMNB2 encodes lamin B2 and lamin B3 protein that arises by alternative RNA splicing. These are expressed in most somatic cells or germ cell-specific, respectively. LMNA encodes lamin A, lamin C, lamin C2, and lamin AΔ10, among which lamin A and lamin C are generated by alternative RNA splicing and expressed in most terminally differentiated cells. On the other hand, lamin C2 is expressed in germ cells, and the expression of lamin AΔ10 is not known (Dechat et al., 2008; Reddy et al., 2008; Worman, 2012).
Lamins not only plays a structural role in nuclear membrane formation, but also regulates chromatin organization and gene expression (Peric-Hupkes et al., 2010; Wilson and Foisner, 2010). However, the exact physiological role in gene expression is not well known.
Fig. 1. The structure of the nuclear envelope.
The inner nuclear membrane (INM) and outer nuclear membrane (ONM) are separated by the perinuclear space (PNS). INM and ONM connect at sites of nuclear pore complex (NPC) where the membrane curves as it surrounds the NPC. The nuclear lamina underlies the nucleoplasmic face of the INM. The ONM is continuous with the endoplasmic reticulum (ER), thus lumen between the ONM and INM is continuous with the ER lumen. INM proteins interact with the nuclear envelope (NE) membrane, chromatin and the lamina. For example, the lamin B receptor (LBR) interacts both with B‑type lamins and chromatin‑associated heterochromatin protein 1 (HP1) in conjunction with core histones. Members of the LEM (lamina‑associated protein 2 (LAP2), Emerin, MAN1)‑domain family bind to lamins and interact with chromatin through barrier‑to‑autointegration factor (BAF). SUN proteins in the INM interact with nesprins in the ONM, thereby forming LINC complexes that establish connections to cytoskeleton in cytoplasm. Figure modified from that of published DATA (Guttinger et al., 2009).
B. Cancer and nuclear shape
In many types of cancer cells, nuclear shape is often abnormal compared with normal cells, making nuclear morphology an indispensable criterion in the current pathological assessment of cancer (Zink et al., 2004; Chow et al., 2012; de Las Heras et al., 2013).
Cancer cells commonly exhibit various aberrations at the genetic and structural levels. Amomg them, defects of the nuclear architecture include nuclear size changes, volume changes, and rupture phenomena. In addition, genome stability and a variety of cellular process are often impaired. Recently, aberrantly expression of proteins that constitute the nuclear envelope and defects of cancer cells has been revealed (de Las Heras et al., 2013). Corresponding changes in the proteins that form the nuclear membrane are also well known and depend on the cancer cell type. Notably, changes in these proteins are being used as cancer biomarkers (Chow et al., 2012).
Interestingly, in ovarian cancer cells, nuclear abnormalities are associated with chromosomal instability and aneuploidy (Capo-chichi et al., 2011). However, it is unclear whether the nuclear abnormalities observed in cancer cells are the cause or consequence of cancer formation and progression. Moreover, how the nuclear abnormalities in cancer cells are formed is not well known.
Since nuclear deformation is a unique feature of cancer cells distinguished from normal cells, I need to clarify the constituents of the nuclear membrane and investigate molecular changes of the nuclear membrane in cancer cells. This is ultimately important to find a biomarker for tumor cell detection and to develope a new cancer therapy strategy.
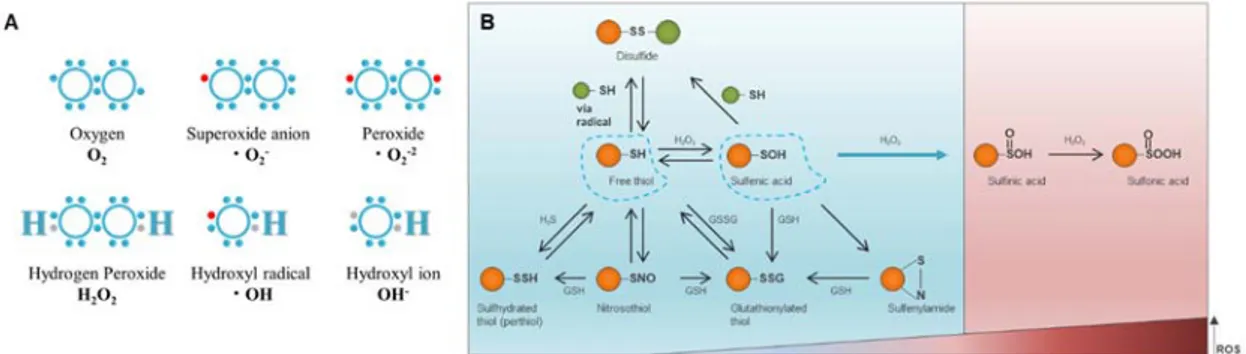
C. Reactive oxygen species (ROS) and oxidative post-translational modification (Ox-PTM)
Most reactive oxygen species (ROS) are free radicals derived from an oxygen molecule which can be categorized into two groups: free oxygen radicals and non-radical ROS. Free oxygen radicals include superoxide (O2•−), hydroxyl radical (•OH), and nitric oxide (NO•).
Non-radical ROS include hydrogen peroxide (H2O2), singlet oxygen (1O2), and
ozone/trioxygen (O3) (Fig. 2A) (Liou and Storz, 2010). ROS can be produced by various intra-
and extracellular factors, and are in balance with intracellular antioxidants such as glutathione. If this balance is broken, free radicals can cause damage to DNA, RNA, proteins, and lipids. It can also cause genetic instabilities or mutations, and can lead to changes in gene expression. All of these consequences of excessive ROS are ultimately capable of producing cancers (Klaunig et al., 2010). In addition, cancer cells, which exhibit an accelerated metabolism, require high ROS concentrations to maintain their high proliferation rate (Pelicano et al., 2004; Sosa et al., 2013). However, the specific relationship between nuclear shape change and ROS in cancer cells is still not well understood.
Covalent modification of proteins is a powerful way to control macromolecular functions and regulates protein activity through post-translational modifications of various methods (Chalker et al., 2009). Among protein amino acids, cysteine has its own unique reactivity (Reddie and Carroll, 2008; Chung et al., 2013). The side chain of the cysteine residue has a terminal thiol (-SH) functional group; the sulfur atom at the thiol center is enriched in electrons and its d-orbitals allow various oxidative post-translational modification (Ox-PTM), such as, -nitrosylation (SNO), sulfhydration (SSH), S-glutathionylation SG), disulfide bonds (RS-SR'), sulfenylation (SOH), sulfinic acid (SO2H), sulfonic acid (SO3H) (Fig. 2B). Thus,
Ox-PTM of the cysteine in the intracellular environment regulates a wide variety of biological phenomena, such as protein stability, catalysis, metal binding, protein turnover, and signal transduction (Holmstrom and Finkel, 2014). The cell cycle is regulated by several types of protein kinases and phosphatases, which are known to modulate their activity through the modification of cysteine residues by oxidative stress, in turn affects the promotion or inhibition of cell proliferation (Chiu and Dawes, 2012).
Fig. 2. Reactive oxygen species and oxidative post-translational modifications (Ox-PTMs) on cysteine.
(A) Electron structures of common reactive oxygen species. (B) Blue is protective mode. From
free thiol (left blue dotted line), modifications induced by small molecules: sulfhydration, S-nitrosylation, S-glutahionylation (bottom) and sulfenylation (right blue dotted line). From sulfenic acid to reversible modifications: disulfide bond formation, sulfenylamide and S-glutathionylation. Red is nonprotective mode, ir- or reversible modification: sulfinic acid and sulfonic acid as reactive oxygen species level increases. Figure modified from that of published DATA (Chung et al., 2013).
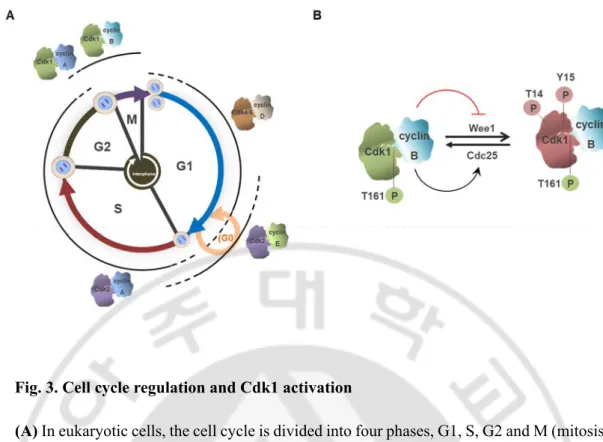
D. Cell cycle and Cyclin-dependent kinases (Cdks)
The cell-division cycle is a series of phenomena that occurs in a cell and is a process of duplicating DNA and dividing it equally into two daughter cells. In eukaryotic cells, the cell cycle is conventionally divided into four phases, G1, S, G2 and M (mitosis), and it has three checkpoints at the onset of S phase, and at entry and exit of mitosis (Morgan, 2007; Hochegger et al., 2008). These checkpoints are regulated by Cyclin-dependent kinases (Cdks), a family of serine/threonine kinases, and each Cdk regulates cell cycle transitions along with their activating cyclin subunits (Fig. 3A) (Lim and Kaldis, 2013; Bai et al., 2017). In most cases, the concentration of the kinase subunit remains constant, while the concentration of the cyclin subunit has the oscillation pattern. Kinases can only be activated by their cyclins, and other processes require phosphorylation or dephosphorylation of specific residues in order to be fully activated.
In animal cells, protein phosphorylation by activation of Cdk1-cyclin B1 plays an important role in the transition of interphase to mitosis (Nigg, 2001; Lindqvist et al., 2009). This kinase promotes the phosphorylation of a wide range of proteins, which results in nuclear envelope breakdown, chromosome condensation, and spindle formation in the cell. Cdk1-cyclin B1 is activated from the G2 phase; first, cyclin B1 recognizes and binds to PSTAIRE helix of Cdk1 (Morgan, 2007; Lim and Kaldis, 2013; Malumbres, 2014), and then T161 present in the activation loop is phosphorylated by Cdk-activating kinase (CAK) (Krek and Nigg, 1992; Solomon, 1994). However, during the interphase, the inhibitory phosphorylation residue of Cdk1, T14 / Y15, is phosphorylated by Wee1 / Myt1 kinase, limiting the activity of Cdk1. During the G2/M transition, Cdk1-cyclin B1 complex is fully activated through the dephosphorylation of inhibitory phosphorylation by Cdc25 phosphatase (Fig. 3B) (Nilsson and Hoffmann, 2000; Kellogg, 2003; Hormanseder et al., 2013).
Fig. 3. Cell cycle regulation and Cdk1 activation
(A) In eukaryotic cells, the cell cycle is divided into four phases, G1, S, G2 and M (mitosis),
each phase is regulated by Cyclin-dependent kinases (Cdks) and their regulatory partner proteins, the cyclins. (B) In mitosis, Cdk1 activity depends on association with cyclin B. After cyclin B associated with Cdk1, Cdk1 activation requires phosphorylation events. First, phosphorylation occurs in the conserved threonine present in the activation loop by CAK (green P, T161). On the other hand, inhibition phosphorylation occurs at residue Thr14/Tyr15 by Wee1/Myt1 kinases (red P, T14, Y15). This inhibitory phosphorylation is dephosphorylated by Cdc25. When initial activation of Cdk1 occurs, the Cdk1-cyclin B complex is more fully activated through phosphorylation of Wee1 and Cdc25, and the cell proceeds to mitosis. Green background Cdk1; inactive form, Red backgound Cdk1; active form.
E. Nuclear envelope disassembly and reassembly during mitosis.
During the interphase of the cell cycle, the nuclear membrane remains intact and relatively stable. However, during mitosis, the nuclear envelope changes dramatically. In early mitosis, the nuclear membrane becomes fragmented, and the nuclear envelope and lamina break down ("disassembly of the nuclear envelope") (Mogessie and Schuh, 2014). Conversely, during mitotic anaphase and telophase, the nuclear structures are gathered around the chromosomes to re-form the nuclear envelope ("reassembly of the nuclear envelope") (Fig. 4) (Hetzer et al., 2005; Kutay and Hetzer, 2008; Guttinger et al., 2009; Schellhaus et al., 2016).
In prophase, NPC disassembly and microtubule-mediated stretching, nuclear lamina tearing and nuclear membrane fragmentation are initiated. During prometaphase, lamina depolymerization and nuclear envelope disassembly occur. In this process, various mitotic kinases play an important role, among which Cdk1-cyclin B complex phosphorylates several proteins in the nuclear membrane, such as NUP, lamins, LBR, and LAP2. In addition, Aurora kinase and Polo-like kinase also play an essential role in mitotic entry, centrosome separation and maturation, and spindle dynamics, thereby controlling the progression of early mitosis (Fig. 4A) (Guttinger et al., 2009; Alvarez-Fernandez and Malumbres, 2014; Mogessie and Schuh, 2014).
In late anaphase, prepores are formed for NPC formation and recruitment of membrane tubules begins. During telophase, INM proteins bind to DNA for NE reformation, NPC is formed, and lamin reassembly occurs (Guttinger et al., 2009; Wandke and Kutay, 2013). Since the kinase plays a crucial role in the nuclear envelope disassembly process, it is important not only to inactivate the kinase but also the phosphatase to reduce the phosphorylation of the substrates in the reassembly process (Fig. 4B). However, phosphatase has not been well studied, recently, it has been reported that protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A) play an important role in the nuclear membrane remodeling process (Vagnarelli et al., 2011; Asencio et al., 2012).
BAF (barrier-to-autointegration factor), the substrate of PP2A, is a protein that binds to the LEM domain of inner nuclear membrane proteins (e.g., LAP2β, Emerin, MAN1) (Haraguchi et al., 2001; Barton et al., 2015) and acts as a bridge between the nuclear envelope and chromatin by interacting with chromatin (Segura-Totten and Wilson, 2004). BAF is phosphorylated by VRK1 (vaccinia related kinase 1) during early mitosis and
dephosphorylated by PP2A during mitotic exit (Asencio et al., 2012; Wandke and Kutay, 2013). Dephosphorylation enables BAF to localize at the chromosome 'core' region, which defines the central region of a set of daughter chromosomes near the spindle pole during telophase (Haraguchi et al., 2008). Emerin, Lap2β and lamin A are recruited to the core region in a BAF-dependent manner during telophase, forming an important structure in the nuclear envelope reassembly process (Haraguchi et al., 2001; Haraguchi et al., 2008; LaJoie and Ullman, 2017). Importantly, it has been reported that knockdown of BAF results in nuclear abnormalities (Furukawa et al., 2003; Gorjanacz et al., 2007; Puente et al., 2011)
Fig. 4. NE disassembly and reassembly.
(A) In prophase, chromatin condensation, formation of microtubule asters around centrosomes
and centrosome separation begins. Microtubules that are attached to the NE lead to NE invaginations around centrosomes and to the formation of holes on the opposing site of the NE. NPC and lamin disassembly initiates and allows for the retraction of NE membranes into the ER. The transition into prometaphase is marked by the loss of the NE permeability barrier. In metaphase, most soluble components of the NE are dispersed throughout the cytoplasm, whereas INM proteins reside in the tubular mitotic ER. (B) In anaphase A, chromatin is compacted, NPC assembly is initiated. During anaphase B, ER membrane tubules start binding to the chromatin surface. During telophase, binding of INM proteins to DNA/chromatin supports the attachment of membrane sheets to chromatin. Lamina is assembled and NPC formation is completed by the step‑wise recruitment of NPC constituents and the NE is sealed. Figure modified from that of published DATA (Guttinger et al., 2009).
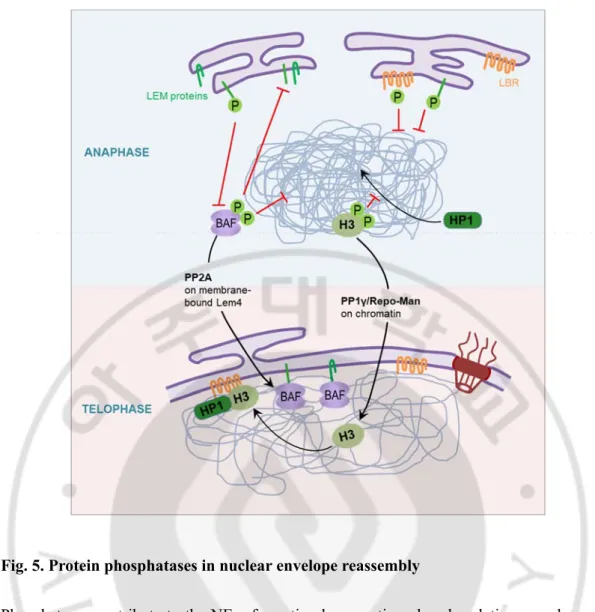
F. Protein phosphatases in mitosis
Mitotic events are spatially and temporally controlled by the balance of various kinases and phosphatases (Wurzenberger and Gerlich, 2011; Mochida and Hunt, 2012; Alvarez-Fernandez and Malumbres, 2014). In particular, the mitotic exit process depends on the removal of mitotic phosphorylation from a broad range of substrates. Thus, mitotic exit regulation involves inactivation of mitotic kinases and activation of counteracting phosphatases. The catalytic subunits of phosphatase are continuously activated, but their overall activity is determined by their intracellular localization, substrate specificity, and their very diverse regulatory subunits. As a result, temporal and spatial activity can be regulated. In budding yeast, Cdc14 is well known as a key phosphatase of the mitotic exit process. However, in animal cells, recently the phosphatases have been identified, including the PP1 and PP2A. PP2A-B55 and PP1 are phosphatases counterbalanced with Cdk1-cyclin B and play a key role in the mitotic exit process (Wandke and Kutay, 2013).
PP1 functions as a heterodimeric complex containing catalytic subunit and regulatory subunit. During mitosis, PP1 regulates chromatin decondensation with Repo-Man and PNUTS, one of the regulatory subunits. (Landsverk et al., 2005; Vagnarelli et al., 2011). PP2A is a heterotrimeric holoenzyme composed of a scaffold A-type subunit, a regulatory B-type subunit, and a catalytic C-type subunit complex. A recent study reported that PP2A-B55 plays an important role in reassembly of the nuclear envelope during mitotic exit (Fig. 5) (Asencio et al., 2012).
Activation of Cdk1 during mitosis inhibits the activity of these two phosphatases. PP1 is inhibited by direct phosphorylation by Cdk1, or by binding to inhibitor 1 protein phosphrylated by Cdk1. PP2A is inhibited by Cdk1-induced Greatwall and ENSA protein phosphorylation. On the other hand, in the mitotic exit process, PP2A maintains its activity by dephosphorylation of the Greatwall, which is activated by Cdk1. Thus, the interaction between Cdk1 and Wee1 / Myt1 kinase and Cdc25, and the interaction of Cdk1 with Greatwall and PP2A controls the phosphorylation of a wide spectrum of substrates and their phosphorylation controls mitotic entry and progression.
Fig. 5. Protein phosphatases in nuclear envelope reassembly
Phosphatases contribute to the NE reformation by reverting phosphorylations on chromatin and NE proteins. The regulatory subunit Repo-Man targets PP1γ to chromatin, causing dephosphorylation of H3. Thus, PP1 controls the association of HP1 with chromatin, contributing to heterochromatin formation. Both HP1 and H3 interact with the INM protein LBR and consequently chromatin bind to the NE. PP2A is recruited to membranes by LEM4, which inhibits the VRK1 (not shown), BAF kinase, and facilitates dephosphorylation of BAF. It allows the interaction of BAF with both chromatin and other LEM domain proteins. Figure modified from that of published DATA (Wandke and Kutay, 2013).
G. Purpose of this study
Nuclear deformation is often observed in many types of cancers (Zink et al., 2004; Chow et al., 2012). In ovarian cancer cells, nuclear envelope structural defects cause chromosomal instability and aneuploidy (Capo-chichi et al., 2011). However, it is unclear whether the nuclear deformation observed in cancer cells is the cause or consequence of cancer formation and progression, and how the nuclear abnormalities in cancer cells are formed is not well-known.
ROS can be produced from endogenous and exogenous sources. Oxidative stress then, in turn, may cause the damage of DNA, proteins, and lipids. It can also cause genetic instabilities or mutations and can lead to changes in gene expression. The resuls of excessive ROS can ultimately produce cancers (Klaunig et al., 2010). Recently, in aging model, it has been reported that H2O2 alters nuclear shape in ataxia-telangiectasia cells by increasing the amount
of lamin B1 protein (Barascu et al., 2012). However, in cancer cells, the specific relationship between nuclear shape change and ROS remains unknown.
During the interphase of the cell cycle, the nuclear membrane remains intact and relatively stable. However, during mitosis, the nuclear envelope undergoes extensive structural reorganization (Kutay and Hetzer, 2008; Guttinger et al., 2009; Schellhaus et al., 2016). Therefore, it is highly likely that the cue that induces abnormal nuclear shape acts in a cell cycle-dependent manner. Specifically, cells in mitosis may be more vulnerable to the events that affect nuclear shape because this is when disassembly and reassembly of nuclear envelopes take place.
Like other mitotic events, nuclear envelope disassembly and reassembly processes are spatially and temporally controlled by the balance of various kinases and phosphatases (Guttinger et al., 2009; Wurzenberger and Gerlich, 2011; Mochida and Hunt, 2012; Alvarez-Fernandez and Malumbres, 2014). A recent study reported that protein phosphatase 2A (PP2A) plays an important role in reassembly of the nuclear envelope during mitotic exit (Asencio et al., 2012). It is well known that PP2A is inhibited by hydrogen peroxide (H2O2) (Rao and
Clayton, 2002; Kim et al., 2003; Foley et al., 2007), and its activity is decreased in various cancer cell types (Grech et al., 2016), suggesting the possible involvement of PP2A in the relationship between ROS and nuclear shape change.
and ROS, which are mainly observed in cancer cells, is dependent on the cell cycle. I assumed that the nuclear shape change by ROS will occur more easily in mitotic cells then asynchronous cells. Moreover, I tried to identify the molecules that contribute to nuclear shape change and reveal the mechanisms that have not been known until now.
In this study, I show that mitotic cells are more vulnerable to ROS than asynchronous cells in terms of abnormal nucleation. As the molecular mechanism, it is suggested that the activity of Cdk1 and PP2A, which are important proteins involved in mitosis, is inhibited by H2O2
treatment. Disruption of Cdk1 activity induces aggregated lamin and occurs premature lamin reassembly during mitosis. In addition, inhibition of PP2A activity results in BAF mislocalization during mitotic exit. Both eventually form abnormal nuclear shape.
II. MATERIALS AND METHODS
A. Antibodies
The following antibodies were used : mouse monoclonal antibodies to α-tubulin (Santa Cruz, sc-23948), BAF (Abnova, H00008815-M07), GAPDH (Santa Cruz, sc-32233), PP2A (BD science, 610555), mAb414 (covance, MMS-120P), GFP (Santa Cruz, sc-9996), HA-probe (Santa Cruz, sc-7392), phospho r-H2A.X (Ser139) (EMD Millipore, 05-636), Lamin A/C (Santa Cruz, sc-7292), cyclin B1 (SantaCruz, sc-245), Cdk1 (SantaCruz, sc-54) at a 1:500 dilution in PBS with 3 % BSA, and Flag (Sigma-Aldrich, F1804) at a 1:5000 dilution in PBS with 3 % BSA; rabbit polyclonal antibodies to lamin B1 (Abcam, ab16048), phospho Cdk1 Y15 (Cell signaling, 9111S), pSerCdks (Cell signaling, 9477S), phospho lamin A/C (Ser22) (Cell signaling, #2026S) at a 1:1000 dilution in PBS with 3 % BSA. Horseradish peroxidase-conjugated mouse (G21040) and rabbit (G21234) antibodies were obtained from Invitrogen (used at a 1:5000 dilution in TBST). The following fluorochrome-conjugated secondary antibodies were used (at a 1:500 dilution in PBS with 3 % BSA): anti-mouse Alexa-488 (Invitrogen, A11059), anti-rabbit Alexa-488 (Invitrogen, A11034), TRITC-conjugated phalloidin (Jackson Immunoresearch, P1951), anti-mouse Cy3 (Jackson Immunoresearch, 715-165-151), Alexa-594 (Invitrogen, A11037).
B. Cell culture
HeLa cells were purchased from American Type Culture Collection (Manassas, VA, USA), and cultured in Dulbecco's modified Eagle's medium nutrient mixture F-12 HAM (DMEM/F12, Sigma-Aldrich, D8900) supplemented with penicillin/streptomycin (Gibco BRL, 15240-062) and 10 % (V/V) fetal bovine serum (Gibco BRL, 16000-044). The hTERT-immortalized retinal pigment epithelial cell line, hTERT RPE-1, was obtained from the ATCC, and cultured in DMEM/F12 supplemented with penicillin/streptomycin, 10 % FBS and 0.01 mg/mL hygromycin B (Sigma-Aldrich, H0654). Human osteosarcoma cell line, U2OS, was also obtained from ATCC, and cultured in DMEM/high glucose medium (Gibco, 31600-034) supplemented with penicillin/streptomycin and 10 % FBS. HT1080 was obtained from ATCC,
and cultured in DMEM (Gibco, 31600-026) supplemented with penicillin/streptomycin and 10 % FBS. All cells were cultured at 37 °C in a humidified incubator with 5 % CO2 in air.
C. Synchronization and drug treatment
Mitotic cell synchronization was performed by thymidine/RO3306 block as described in the previous paper (Cho et al., 2017). Briefly, cells (5×105) were seeded into 100 mm culture plates
(BD FALCON, 353003), cultured in DMEM/F12 or DMEM containing 10 % FBS for 1 day and subsequently treated with 1 mM (for HeLa cells) or 2.5 mM thymidine (for U2OS, RPE-1, and HT1080 cells) thymidine (Sigma-Aldrich, T9250) to arrest at G1/S border. Twenty hours later, cells were washed with thymidine-free medium and cultured in complete medium for 7 h (for HeLa cells) or 8 h (for U2OS, RPE-1, and HT1080 cells). Then, cells were cultured again in 9 μM RO3306 (Enzo, ALX-270_463) containing medium to arrest at G2/M border for 2 h (for HeLa cells) or 3 h (for U2OS, RPE-1, and HT1080 cells). Mitotic cells were isolated by mechanical shake-off following 30 min release from RO3306 treatment. N-Acetyl-L-cysteine (NAC, A9165, Sigma-Aldrich, U.S.A) was treated at the final concentration of 10 mM at 30 min before H2O2 treatment, unless otherwise stated.
D. H2O2 treatment
Ten mM H2O2 (Sigma-Aldrich, 216763) solution was prepared in Distilled Water
immediately before use, and then treated to culture media to the final concentration of 50, 100 or 200 μM. The actual concentration of H2O2 was determined by measuring OD 240 nm using
the molar extinction coefficient of 43.6 M−1 cm−1 (Noble and Gibson, 1970).
E. Determination of intracellular ROS level
HeLa Cells (1×105) were plated on a 60 mm culture plate (Techno Plastic Products,
TP93060) and subsequently treated with 2 mM thymidine to arrest at G1/S border. Twenty hours later, cells were washed with thymidine-free medium and cultured in complete medium for 8 h. Then, cells were cultured again in 100 ng/ml nocodazole (Sigma, M1404) containing medium to arrest at mitosis for 3 h. Mitotic cells were isolated by mechanical shake-off. N-Acetyl-L-cysteine (NAC) was treated at the final concentration of 10 mM at 30 min before
H2O2 treatment. Then, cells were exposed to hydrogen peroxide solution during 1 h and then
cells were washed with phosphate- buffered saline (PBS). Cells were then treated with 20 μM dichlorodihydrofluorescein diacetate (DCF-DA) for 20 min. The amounts of intracellular ROS were determined as the fluorescence intensities of DCF-DA with a BD FACSAria™ III using an Argon laser with 525 nm (DCF-DA) band pass filter. Data were analyzed with WIN MDI software (Windows multiple document interface for flow cytometry: http://facs.scripps.edu/).
F. Measurements of intracellular level of H2O2 in living cells
The intracellular level of H2O2 was measured using a fluorescent sensor pHyper-Cyto
(Belousov et al., 2006), and the experimental procedure was described in the previous paper (Cho et al., 2017). Briefly, two days before experiment, HeLa cells were transfected with pHyper-Cyto using electroporation, and the fluorescence was monitored by using a microscope (Nikon eclipse Ti with a 20X14 NA Plan-Apochromat objective). Images were captured with an iXonEM +897 Electron. pHyper has two excitation peaks with maxima at 405 and 488 nm, and one emission peak. Upon reaction with H2O2, the excitation peak at 405
nm decreases proportionally to the increase in the peak at 488 nm (Belousov et al., 2006). Therefore, the change of intracellular H2O2 level could be evaluated by calculating the pHyper
fluorescence ratio upon excitation by 488 and 405 nm laser. pHyper fluorescence was acquired in two different channels: CH1 (excitation 488 nm) and CH2 (excitation 405 nm) in time-lapse imaging. The images are expressed as the ratio of CH1 to CH2. Images were taken at the interval of 30 s for 25 min. A higher index corresponds to higher levels of intracellular H2O2.
The image analysis was performed in NIS elements Ar microscope imaging software.
G. Plasmids and transfection experiment
HA-catalase or GFP-BAF and -Emerin, Flag-PP4 was a gift from Dr. Gyesoon Yoon (Ajou University, Korea) or Dr. Yasushi Hiraoka (Osaka University, Japan) or Daniel Durocher (University of Toronto), respectively. HA-Cdk1 was supported from Dr. Chang-Woo Lee (Sungkyunkwan University, Korea). CFP-PP2A was gifted from Dr. Hyeseong Cho (Ajou University, Korea), which was subsequently subcloned into Flag-destination or emGFP-destination vector to generate Flag-PP2A and emGFP-PP2A, respectively, using gateway
system. GFP-lamin B1was a gift from Iain W. Mattaj (European Molecular Biology Laboratory, Germany) and 3A mutant was constructed using a Muta-DirectTM Site Directed
Mutagenesis Kit (iNtRON BIOTECHNOLOGY, #15071). GFP-Sec61β (plasmid #15108) and GFP-PP1 (plasmid #44224) were purchased from addgene. HeLa cells were transfected using neon electroporation (Invitrogen).
H. Transmission electron microscopy
Cells were prefixed in Karnovsky’s solution (1 % paraformaldehyde, 2 % glutaraldehyde, 2 mM calcium chloride, 0.1 M cacodylate buffer, pH 7.4) for 2 h and washed with cacodylate buffer. Post-fixing was carried out in 1 % osmium tetroxide and 1.5 % potassium ferrocyanide for 1 h. After dehydration with 50–100 % alcohol, the cells were embedded in Poly/Bed 812 resin (Pelco, Redding, CA, USA), polymerized and observed under electron microscope (TEM; Zeiss EM 902 A, Zeiss, Oberkochen, Germany).
I. Immunocytochemistry
Mitotic cells were split onto poly-L lysine (PLL, P6282, Sigma-Aldrich)-coated slides. Cells were grown on coverslips, and fixed in 4 % formaldehyde or 10 % trichloroacetic acid (for analysis of BAF localization) in PBS for 15 min at RT and permeabilized with 0.2 % Triton X-100 (Sigma, 9002-93-1) in PBS for 10 min at RT. Fixed cells were pre-incubated in blocking solution (3 % bovine serum albumin in PBS), followed by incubation with primary antibodies at 4 °C for overnight. After incubation with primary antibodies, cells were washed three times with shaking in PBS, and probed with fluorescein (Cy3, Alexa 488, Alexa 549) – conjugated anti-mouse or anti-rabbit secondary antibodies. After washing three times with PBS, DAPI (Invitrogen, D3571) was used for DNA counterstaining. Three times of washing with PBS was followed by mounting in the mounting solution (Biomeda, M01). Samples were examined by a fluorescence microscope (Axio Imager M1, Carl Zeiss).
J. Immunoblotting
Conventional immunoblotting was performed as previously described (Nam et al., 2008) using corresponding antibodies. Briefly, cell lysates (30 μg) were resolved by sodium dodecyl
sulfate–polyacrylamide gel electrophoresis and then were transferred to polyvinylidene fluoride (PVDF) membrane. After blocking for 1 h at room temperature (RT) with TBS containing 0.1 % (V/V) Tween‐20 (TBST) and 5 % (W/V) non-fat milk, membranes were incubated with corresponding primary antibodies at 4 °C, followed by washing with TBST and incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody (Amersham Biosciences, Piscataway, NJ) for 1 h at RT. Detection was carried out using ECL reagents (Amersham Biosciences, RPN2106) and exposing them to x-ray film.
K. Protein Phosphatase 2A (PP2A) activity assay
Phosphatase activity was determined using the DuoIC set PP2A phosphatase activity kit (R&D Systems, DYC3309-2) according to the manufacturer's instructions. Cells were rinsed two times with TBS. Cells were solubilized in 1 ml of lysis buffer (50 mM HEPES, 0.1 mM EGTA, 0.1 mM EDTA, 120 mM NaCl, 0.5 % Nonidet P-40, pH 7.5, 25 μg/ml leupeptin, 25 μg/ml pepstatin, 2 μg/ml aprotinin, 1 mM PMSF)/1 × 107 cells. Cell extract was centrifuged at
2000 × g for 5 min, and sample protein concentration was quantified using Bradford assay. Three hundred μg of the cell lysate was added to 96-well plates coated with immobilized capture antibody specific for the catalytic subunit of PP2A. After removing unbound material, a serine/threonine synthetic phosphopeptide substrate, which is dephosphorylated by active PP2A to generate free phosphate and unphosphorylated peptide, was added. The free phosphate released during the 30-min incubation was then detected by a dye binding assay using malachite green and molybdic acid. The activity of PP2A was determined by calculating the rate of phosphate release.
L. Time-lapse microscopic analysis
For time-lapse live cell imaging, HeLa cells were transfected with GFP-BAF or EGFP-lamin B1, and cells were (1×104) seeded onto a 4-well Glass-based dish (Thermo Scientific™
Nunc™Lab-Tek II Chambered* Coverglass, 154526). Cells were treated with 2 mM thymidine (Sigma-Aldrich, T9250) to arrest at G1/S border. Twenty h later, cells were washed with thymidine-free medium and cultured in complete medium for 7 h. Then, cells were cultured again in 9 μM RO3306 (Enzo, ALX-270_463) containing medium to arrest at G2/M
border for 2 h. Cells were released from RO3306 treatment, and stained with Hoechst 33342 for visualization of chromosomes. After 30min, cells were treated with 50 μM H2O2 or 100
nM okadaic acid. Fluorescence images were acquired every 3 min using a Nikon eclipse Ti with a 20X14 NA Plan- Apochromat objective. Images were captured with an iXonEM +897 Electron Multiplying charge-coupled device camera and analyzed using NIS elements Ar microscope imaging software.
M. In vitro Cdk1 kinase assay
HeLa cells were treated with 1mM thymidine to arrest at G1/S border. Twenty hours later, cells were washed with thymidine-free medium and cultured in complete medium for 6 h. Then, cells were cultured again in 100 ng/ml nocodazole (Signa, M1404) containing medium to arrest at mitosis for 4 h. Mitotic cells were isolated by mechanical shake-off. 300 μM H2O2
or 9 μM RO3306 was treated for 1h. Mitotic cells were lysed with IP buffer (50 mM Tris-HCl, 150 mM NaCl, 0.5 % NP-40, 5 mM EDTA), 400 μg of cell lysates were incubated with 1 μg of cyclin B1 antibody at 4 °C for overnight. Each tube was incubated with 30 μl Protein G (GE, 17-0618-01) beads at 4 °C for 1 h. The precipitates were washed three times with lysis buffer and twice with kinase buffer (50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 1 mM DTT, 5
mM β-glycerolphosphate). Cdk1 kinase assay on histone H1 (SantaCruz, sc-221729) was performed by mixing the respective immune complexes with 1 μg of histone H1 and 10 μCi of [γ-32P] ATP (Perkin Elmer, #NEG002A250UC) in 30 μl of kinase buffer. The reaction
mixtures were resolved by SDS-PAGE analysis. Gels were stained with Coomassie Blue staining solution and dried. Phosphorylated substrates were detected by autoradiography.
N. Immunoprecipitation (IP)
For immunoprecipitation of Cdk1, cells were transfected with HA-Cdk1 plasmid, early mitotic cells obtained by shake off were arrested for prometaphase by treatment with nocodazole. Cells treated with H2O2 depending on concentration for 1 h, and cells were lysed
with IP buffer (50 mM Tris-HCl, 150 mM NaCl, 0.5 % NP-40, 5 mM EDTA), 400 μg of cell lysates were incubated with 1 μg of HA-antibody at 4 °C for overnight. Each tube was incubated with 30 μl Protein G (GE, 17-0618-01) beads at 4 °C for 1 h. After three times of
washing with IP buffer, beads were boiled with 30 μl non-reducing sample buffer for 5 min. The samples were separated by SDS-PAGE and then were transferred to PVDF membrane. After blocking for 1 h at RT with TBST and 5 % (W/V) non-fat milk, the membranes were incubated with indicated antibodies at 4 °C for overnight. After three times washing with TBST, the membranes were incubated with secondary antibody (TrueBlot; ROCKLAND, 18-8817-30) at room temperature for 1 h. Detection was carried out using ECL reagents (Amersham Biosciences, RPN2106) and by exposing them to X-ray film.
O. Phos-tag gel analysis
The phos-tag reagents were purchased from Wako Chemicals, and gels containing phos-tag were prepared according to the manufacture’s instructions. For immunoprecipitation, cells were lysed with mild lysis buffer (50 mM HEPES at pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 10 mM pyrophosphate, 10 mM glycerophosphate, 50 mM NaF, 1.5 mM Na3VO4, 1
mM PMSF, 1 mM DTT, protease inhibitor cocktail (Roche)), and centrifuged at 12,000 × g for 15 min at 4 °C. The supernatants were incubated with the appropriate antibodies for 2 h at 4 °C, and protein G or protein A conjugated beads were added in for additional 1 h. Immunoprecipitates were collected by centrifugation and washed four times with lysis buffer, and proteins were eluted with SDS-PAGE sample buffer.
P. S-glutathionylation assay
Asynchronous or mitotic HeLa cells were preincubated for 1 h with culture medium containing 250 μM biotinylated glutathione ethyl ester (BioGEE) (Invitrogen, G36000) and subsequently exposed to H2O2 for 30 min in a concentration dependent manner. Cells were
washed with ice-cold PBS and lysed in a IP buffer containing 10 mM N-ethylmaleimide (NEM) to block further thiol oxidation. 500 μg of cell lysates were incubated for 1 h at 4 °C with 50 μl streptavidin-conjugated agarose beads (Invitrogen, 88816). Then beads were rinsed three times with IP buffer, S-glutathionylated proteins were released from the beads by boiling with 30 μl sample buffer for 5 min. The samples were separated by SDS-PAGE, and Cdk1 levels were evaluated by Western blot analysis.
Q. Statistical analysis
Most data are represented as mean ± standard deviation (SD). Each experiment was performed in triplicates. Statistical differences were analyzed by Student's t-test and the asterisk (*) and sharp (#) indicates a significant difference *,#; P<0.05, **,##;, P<0.01, ***,###;,
III. Result
PART Ⅰ. Inhibition of PP2A activity by H
2O
2during mitosis disrupts nuclear
envelope reassembly and alters nuclear shape.
A. Treatment of mitotic cells with H2O2 induces abnormal nucleation.
To determine whether the cue that induces abnormal nuclear shape functions in a cell cycle-dependent manner—specifically, whether mitosis is a sensitive period for the formation of abnormal nuclear shape—I compared the effects of H2O2 treatment on asynchronous and
mitotic cells, obtained following the scheme shown in Fig. 6A. After 10 h of H2O2 treatment,
which provides sufficient time for mitotic cells to enter the next interphase during which altered nuclear morphology is observed, I monitored changes in nuclear shape by immunostaining for lamin B1. Treatment of asynchronous cells with H2O2 had little effect on
nuclear shape in most cells except at high concentrations of H2O2 with longer treatment
duration. In contrast, treatment with H2O2 during mitosis caused marked,
concentration-dependent changes in nuclear shape in the subsequent interphase, inducing significant changes at an H2O2 concentration of 50 μM and reaching a plateau at 100 μM; in both cases, nuclear
shape was analyzed at 10 and 24 h after H2O2 treatment. Indeed, mitotic cells showed a
significantly higher tendency to form abnormal nuclear shapes than asynchronous cells under every H2O2 treatment condition (Fig. 6B). Notably, neither 50 nor 100 μM H2O2,
concentrations that are easily achievable in a pathological setting (e.g., a rat ischemia/reperfusion model (Hyslop et al., 1995)), caused cell death after 24 h, as our lab reported previously (Cho et al., 2017).
Treatment of mitotic cells with H2O2 was followed by a variety of changes in nuclear shape,
including folding or fragmentation of the nuclear envelope or adoption of a globular shape. This was also confirmed in simulated 3-dimensional (i.e., 2.5D) images (Fig. 6C). Furthermore, electron microscopy revealed that the nuclear envelope in cells treated with H2O2 during
mitosis formed a curved section with electron-dense sites that may indicate thickening of the nuclear membrane (Fig. 6D).
To measure the abnormal nuclear shape more objectively, the extent of the variability in lamin B1 staining intensity was analyzed. Since the intensity of lamin B1 staining in folded or curved nucleus is more variable than that of normally shaped nucleus, I reasoned that the standard deviation of these values would be an indicator of the degree of nuclear shape alteration. Consistent with the result of counts of abnormal nuclei, lamin B1 staining in mitotic cells exhibited a significantly larger standard deviation than that in asynchronous cells both 10 and 24 h after H2O2 treatment (Fig. 6E). The circularity of the nucleus was quantified as
another approach for objectively representing changes in nuclear shape (Fig. 6F). A circularity value of “1” corresponds to a complete circle, whereas smaller values denote greater deviations from circularity. As a reference point, the mean circularity values of control asynchronous and mitotic HeLa cells were both ~0.8. Whereas the mean circularity value at 10 and 24 h after H2O2 treatment of asynchronous cells was maintained at ~0.8 regardless of the concentration
Fig. 6. H2O2 treatment to mitotic cells forms more abnormal nuclear shape than
asynchronous cells. (A) Experimental design to obtain mitotic cells. (B) Left panel:
Asynchronous (upper) or mitotic (lower) HeLa cells were treated with 100 μM H2O2 for 10 h
and subjected to immunocytochemistry for lamin B1 (green), α-tubulin (red) and DAPI (blue). Scale bar: 20 μm. Right panel: Asynchronous or mitotic cells were treated with H2O2 at
indicated concentrations, and percentage of cells with abnormal nuclear shape was measured after 10 h or 24 h. Results are shown as the mean ± SD from three independent experiments (n=300), *; Control versus H2O2, #; Asynchronous versus Mitosis, *p<0.05, **, ##p<0.01, ###p<0.001 by Student's t-test. (C) Representative examples of abnormal nuclear shape in
H2O2-treated cells. Upper panel; Lamin B1 staining (green). Lower panel; Images from upper
panels were converted to 2.5 dimensional images by using ZEISS Microscope software ZEN. Scale bar: 5 μm. (D) Electron microscopy images of nuclear envelope in cells treated with or without H2O2 for 10 h. Scale bar: 2.5 μm. (E) Standard deviation of lamin B1 intensity inside
an imaginary circle in the nucleus was measured by using ZEN software with the same samples in (B). Results are shown as the mean ± SD (n=50), *; Control versus H2O2, #; Asynchronous
versus Mitosis, #p<0.05, **, ##p<0.01, ###p<0.001 by Student t-test. (F) Nuclear circularity was
calculated by using ImageJ software with the same samples in (B). Results are shown as the mean ± SD (n=30). *; Control versus H2O2, #; Asynchronous versus Mitosis, #p<0.05, **, ##p<0.01, ###p<0.001 by Student's t-test.
To determine whether the effects of H2O2 on the cell cycle were limited to
continuous-exposure conditions, I also tested the effects of transient continuous-exposure to H2O2. Treatment with
H2O2 for 2 h followed by wash-out produced the same susceptibility of mitotic cells to
abnormal nucleation compared with asynchronous cells, as shown by measuring the variability of lamin B1 immunostaining intensity and assessing the circularity index (Fig. 6G–I). This enhanced vulnerability of mitotic cells to abnormal nucleation following H2O2 treatment
compared with asynchronous cells was observed not only in HeLa cells, but also in U2OS, RPE-1 and HT1080 cells, indicating the generalizability of my observations (Fig. 6J).
Fig. 6. H2O2 treatment to mitotic cells forms more abnormal nuclear shape than
asynchronous cells. (G) Asynchronous or mitotic cells were treated with H2O2 at indicated
concentrations. Two hours later, cells were washed out, and the percentage of cells with abnormal nuclear shape was determined 10 h or 24 h after H2O2 treatment. Results are shown
as the mean ± SD from three independent experiments (n=300), *; Control versus H2O2, #;
Asynchronous versus Mitosis, *p<0.05, **, ##p<0.01, ###p<0.001 by Student's t-test. (H)
Standard deviation of lamin B1 intensity inside an imaginary circle in the nucleus was measured by using ZEN software with the same samples in (G). Results are shown as the mean ± SD (n=50), *; Control versus H2O2, #; Asynchronous versus Mitosis, #p<0.05, **, ##p<0.01, ###p<0.001 by Student's t-test. (I) Nuclear circularity was analyzed with ImageJ with the same
samples in (G). Results are shown as the mean (n=30). *; Control versus H2O2, #;
Asynchronous versus Mitosis, *p<0.05, **, ##p<0.01, ###p<0.001 by Student's t-test. (J)
Mitotic U2OS, RPE-1 and HT1080 cells were treated with H2O2 at indicated concentrations,
and percentage of cells with abnormal nuclear shape was determined after 10 h. Results are shown as the mean ± SD from three independent experiments (n=300), *p<0.05, **p<0.01, ***p<0.001 by Student’s t-test.
B. Formation of abnormal nuclei following H2O2 treatment is prevented by NAC or
catalase.
To confirm that the formation of abnormal nuclear shapes in H2O2-treated mitotic cells was
actually caused by ROS, I pretreated cells with the antioxidant N-acetyl-L-cysteine (NAC). Indeed, changes in nuclear shape after H2O2 treatment were almost totally prevented by NAC
(Fig. 7A). The ROS-lowering effect of NAC on mitotic cells under these treatment conditions was verified by fluorescence-activated cell sorting (FACS) analysis using the fluorescent ROS indicator, dichlorodihydrofluorescein diacetate (DCF-DA) (Fig. 7B).
To more specifically address whether H2O2 was responsible for the abnormal nucleation,
prior to H2O2 treatment, catalase, an enzyme that converts H2O2 to water and oxygen, was
transfected into the cells. Whereas cells that did not express catalase exhibited a change in nuclear shape, as expected, nuclear shape change was remarkably reduced in cells overexpressing catalase (Fig. 7C). Interestingly, the formation of abnormal nuclei was also suppressed in catalase-expressing cells in the absence of H2O2 treatment, suggesting that basal
levels of H2O2 induce formation of a basal level of abnormal nuclei. The H2O2-lowering effect
of ectopically expressed catalase was confirmed using pHyper-Cyto (Fig. 7D), a specific fluorescent protein probe for H2O2 (Belousov et al., 2006). Collectively, these findings indicate
that mitotic cells are more prone to abnormal nucleus formation following H2O2 treatment than
asynchronous cells, and that this phenomenon is directly attributable to ROS, based on the preventive effect of NAC treatment and catalase overexpression.
Fig. 7. Nuclear shape alteration induced by H2O2 treatment during mitosis is rescued by
antioxidants. (A) N-acetyl-L-cysteine (NAC) was pretreated (NAC+H2O2) or not (H2O2) for
30 min. Then, mitotic cells were obtained through shake-off, and treated with 100 μM H2O2
for 10 h. Left panel; After 10 h, the nuclear shape was observed by lamin B1 staining (red). Scale bar: 20 μm. Right panel; percentage of the cells with abnormal nuclear shape. Results are shown as the mean ± SD from three independent experiments (n=300), *p<0.05, **p<0.01 by Student's t-test. (B) Mitotic cells were pretreated with NAC or not for 30 min, and then incubated with 100 μM H2O2 for1h. Intracellular ROS level was measured by FACS analysis
using DCF-DA. (C) HeLa cells were transfected with an HA-catalase expressing vector, and then mitotic cells were treated with or without 100 μM H2O2 for 10 h. Left panel; nuclear shape
of HA-catalase (red)- transfected cells. Scale bar: 20 μm. Right panel; percentage of the cells with abnormal nuclear shape according to HA-catalase expression. Results are shown as the mean ± SD from three independent experiments (n=100), *p<0.05, **p<0.01, ***p<0.001 by Student's t-test. (D) HeLa cells were transfected with pHyper-Cyto, and then with pcDNA3 or HA-Catalase. Changes in intracellular H2O2 level is expressed as the ratio upon excitation by
488 and 405 nm laser. Representative time-lapse fluorescence images (30 s interval for 25 min) of pHyper-cyto sensors in 100 μM H2O2-treated mitotic HeLa cells.
C. Quantitative changes in lamin B1, structural changes in the cytoskeleton and ER, and DNA damage are not major contributors to H2O2-induced nuclear shape
changes.
Previous studies have reported that p38 MAPK (mitogen-activated protein kinase) is activated by ROS in ataxia-telangiectasia cells, and that the level of endogenous lamin B1 is increased in these cells, resulting in nuclear deformation and senescence (Barascu et al., 2012). Therefore, I investigated whether the formation of abnormal nuclei in response to H2O2
exposure under my experimental conditions was accompanied by changes in the level of lamin B1. Lamin B1 levels were not noticeably changed at 10 or 24 h after treatment with H2O2 (Fig.
8A). And, similar results were obtained when the cells were transiently treated with H2O2 (Fig.
8B), excluding the possibility that quantitative changes in lamin B1 levels are involved in H2O2-induced abnormal nucleation.
The nucleus is connected directly or indirectly with cytoskeletal elements, including actin filaments, microtubules and intermediate filaments, and these physical connections are known to determine nuclear shape by creating tension between the nucleus and the cytoskeleton (Wang et al., 2009; Lombardi et al., 2011; Jevtic et al., 2014). To address whether alterations in nuclear shape were induced by changes in the cytoskeleton, mitotic cells were treated with H2O2 and assessed the appearance of the cytoskeleton under a microscope. These experiments
showed no significant changes in the appearances of F-actin or microtubules (Fig. 8C). I also investigated possible structural changes in the ER, which is connected to the outer nuclear membrane and thus it could affect nuclear shape (Guttinger et al., 2009; Alvarez-Fernandez and Malumbres, 2014). Changes in ER shape were observed by monitoring ectopically expressed GFP-Sec61β, a fluorescently tagged ER membrane protein, following treatment of mitotic cells with H2O2. These experiments revealed no significant changes in ER structure
(Fig. 8D). Taken together, these results show that the changes in nuclear shape induced by H2O2 were not accompanied by quantitative changes in lamin B1, or structural changes in the
cytoskeleton or ER. In contrast, I did detect changes in the immunocytochemical images of other elements that constitute the nuclear membrane, such as lamin A/C, emerin, and the NPC in response to increases in ROS during mitosis that were associated with changes in nuclear shape (Fig. 8E–G).
et al., 2007), and our group has previously reported that H2O2 induces DNA damage and
subsequent chromatin bridge formation in mitotic cells, changes that appear to be related to binucleation (Cho et al., 2017). To determine whether DNA damage is involved in abnormal nucleus formation, I compared the effects of H2O2 and etoposide, an inhibitor of topoisomerase
II that induces DNA double-strand breaks. Notably, treatment with a high concentration of etoposide induced an increase in the number of cells with abnormal nuclei, suggesting that DNA damage does contribute to the formation of an abnormal nucleus. However, although expression of the DNA damage marker, γ-H2A.X, was increased to a greater extent by 10 μM etoposide than by 50 μM H2O2, the percentage of cells that formed abnormal nuclei in response
to 10 μM etoposide was significantly less than that in response to 50 μM H2O2. A comparison
of 40 μM etoposide and 100 μM H2O2 also showed the same tendency (Fig. 8H), indicating
that DNA damage plays at most a modest role in the formation of abnormal nuclei. Therefore, mechanisms other than DNA damage appear to be of primary importance in the nuclear shape changes induced by H2O2.
Fig. 8. H2O2 - induced abnormal nuclear shape is mainly neither due to changes in lamin
B1 level, nor in cytoskeleton network, nor in DNA damage. (A) Asynchronous or mitotic
HeLa cells were treated with H2O2 at indicated concentrations for 10 h or 24 h. Cell lysates
were harvested and subjected to western blot analysis by using indicated antibodies. (B) Asynchronous or mitotic HeLa cells were treated with H2O2 at indicated concentrations. Two
hours later, cells were washed out. Cell lysates were harvested 10 h or 24 h after H2O2 treatment,
and subjected to western blot analysis by using indicated antibodies. (C) Mitotic HeLa cells were treated with 100 μM H2O2 for 10 h, and cytoskeleton fibers were detected by
immunocytochemistry. Actin filaments and microtubules were visualized by using Phalloidin-TRITC (red) and α-tubulin (red) antibody, respectively. Lamin B1 (green), DAPI (blue). Scale bar: 30 μm. Dotted boxes were magnified as shown. (D and E) HeLa cells were transfected with expression vectors of GFP-Sec61β (D), an ER membrane protein, or GFP-Emerin (E), an inner nuclear membrane protein, and then mitotic cells were treated with 100 μM H2O2 for 10
h. Structure of ER and nuclear envelope were visualized by using GFP (green) antibody. Lamin B1 (red), DAPI (blue). Scale bar: 20 μm. (F and G) Mitotic HeLa cells were treated with 100 μM H2O2 for 10 h, and structure of the nuclear envelope was assessed. Lamin A/C (F) and
nuclear pore complexs (G) were visualized by using lamin A/C (green) and mAb414 (red) antibody, respectively. α-tubulin (red), lamin B1 (green), DAPI (blue). Scale bar: 10 μm. (H) Mitotic HeLa cells were treated with H2O2 or etoposide at indicated concentrations. Upper
panel; Mitotic cells were co-treated with H2O2 or etoposide and 100ng/ml nocodazole,
respectively, for 1 h, and subjected to western blot analysis by using indicated antibodies. Lower panel; Mitotic HeLa cells were treated with H2O2 or etoposide for 10 h, and then
percentage of the cells with abnormal nuclear shape was determined. Results are shown as the mean ± SD from three independent experiments (n=300). ***p<0.001 by Student's t-test. S.E; short exposure, L.E; long exposure.
D. H2O2 inhibits PP2A activity during mitosis.
Since nuclear envelope disassembly and reassembly occur during mitotic entry and exit, respectively, I hypothesized that the observed propensity for mitotic cells to undergo changes in nuclear shape in response to H2O2 was attributable to effects of H2O2 on nuclear envelope
disassembly and/or reassembly processes. It has previously been shown that PP2A plays an important role in the nuclear envelope reassembly process during mitotic exit (Asencio et al., 2012). To determine whether PP2A is involved in the formation of abnormal nuclei in my experimental system, I investigated changes in nuclear shape following treatment of mitotic cells with different concentrations of the PP2A inhibitor, okadaic acid. Although both PP1 and PP2A are inhibited by okadaic acid, it has been reported that PP2A is more sensitive to okadaic acid (in vitro IC50 ≈ 0.5 nM) than PP1 (IC50 ≈ 42 nM) (Foley et al., 2007; Zhuang et al., 2014).
Treatment of mitotic cells with okadaic acid for 2 h caused robust, concentration-dependent changes in nuclear shape, affecting ~94% of cells at the highest concentration tested (150 nM); by contrast, okadaic acid had little effect on nuclear shape in asynchronous cells at any concentration (Fig. 9A). Thus, inhibition of PP2A results in abnormal nucleus formation, but only when applied during mitosis, a phenomenon comparable to the observed greater vulnerability of mitotic cells to H2O2-induced nuclear shape changes. Since H2O2 is known to
decrease PP2A activity in asynchronous cells (Rao and Clayton, 2002; Kim et al., 2003; Foley et al., 2007), I investigated whether PP2A activity was also reduced by H2O2 in mitotic cells
(Fig. 9B). In vitro PP2A activity was assayed after treatment of mitotic cells with H2O2 or
okadaic acid for different durations. Indeed, both H2O2 and okadaic acid decreased the activity
of PP2A in mitotic cells. In addition, the H2O2-induced decrease in PP2A activity was found
to be dependent on H2O2 concentration, and showed a tendency towards recovery in cells
treated with NAC (Fig. 9C). Therefore, H2O2 inhibits the activity of PP2A in mitotic cells,
potentially affecting the nuclear envelope reassembly process and causing changes in nuclear shape.