Hepatitis B Virus X Protein Modulates
Transcriptional Activity of PPAR
γ
through Direct Protein-Protein Interaction
Youn-Hee Choi
Department of Medical Science
The Graduate School, Yonsei University
Hepatitis B Virus X Protein Modulates
Transcriptional Activity of PPAR
γ
through Direct Protein-Protein Interaction
Directed by Professor Se Jong Kim
A Doctorial Dissertation Submitted to
the Department of Medical Science,
the Graduate School, Yonsei University
in partial fulfillment of the requirements
for the degree of Doctor of Philosophy
Youn-Hee Choi
December 2002
This certifies that the dissertation of
Youn-Hee Choi is approved.
_______________________________________
Thesis Supervisor
_______________________________________
Thesis Committee Member #1
_______________________________________
Thesis Committee Member #2
_______________________________________
Thesis Committee Member #3
_______________________________________
Thesis Committee Member #4
The Graduate School
Yonsei University
감사의 글
항상 자상한 배려와 관심으로 지켜봐 주시고 지도해주신 김세종 교수님과 부족한 저에게 자율적으로 연구하고 배울 수 있는 기회를 주시고 아낌없는 배려로 지도해주신 박전한 교수님께 진심으로 감사드립니다. 또한, 많은 조 언으로 논문을 지도해주신 김종선 교수님, 박영년 교수님, 성제경 교수님께 깊은 감사를 드립니다. 늘 격려해주신 최인홍 교수님과 많은 가르침을 주 신 이원영 교수님, 이봉기 교수님, 조상래 교수님, 신전수 교수님께 감사드 립니다. 또한 세종대 이미옥 교수님께도 감사의 글을 올립니다. 저에게 좋은 선배로서 실험실 생활의 열정을 보여주신 신의철 선생님과 오랜시간 함께하고 많은 도움을 준 박상면 선생님께 감사드리며, 항상 곁 에서 도움과 용기를 준 최유정 선생님, 실험에 힘을 더해준 고시환 선생님 과 정한영 선생님께도 감사를 드립니다. 늘 도와주고 성원해준 김혜미 선 생님과 최경선 선생님을 비롯한 많은 선후배들과 미생물학 교실원 여러분 들께 깊은 감사를 드립니다. 오늘의 제가 있게 하신 하느님께 감사드리며, 부족한 저를 이해해주시고 끝없는 성원을 보내주시는 시부모님과 깊은 사랑으로 항상 저에게 힘이 되 어 주시는 부모님께 진심으로 감사드립니다. 어린 동생을 각별한 애정으로 이끌어주는 오빠와 새언니, 힘든 일들 함께해준 동생부부께도 깊은 감사를 드립니다. 마지막으로, 모든 어려움을 함께 해주고 굳은 믿음으로 항상 든든한 버 팀목이 되어준 남편 국경훈과 사랑하는 딸 윤아에게 이 논문을 바칩니다.Contents
List of Figures
Abstract ...1
I. Introduction...3
II. Materials and methods ...9
1. Cell lines and culture...9
2. Plasmids and plasmids construction...9
3. HBx-TG mice...10
4. Antibodies and reagents ...11
5. Immunoprecipitation and Western blot analysis ...11
6. In vitro binding assay and native gel shift assay ...12
7. Immunofluorescent assay (IFA) ...13
8. Northern blot analysis ...14
9. Electromobility shift assay (EMSA) ...14
10. Luciferase reporter assay...15
11. Cytotoxicity assay ...16
III. Results ...18
1. HBx Binds to PPARγ in vivo and in vitro...18
2. HBx Alters the Subcellular Localization of PPARγ. ...22
3. Expression of PPARγ in the HBx-TG Mouse Liver Is Reduced in the Nucleus. ...24
4. The DNA Binding of PPARγ Is Reduced in HBx-TG Mouse...26
5. HBx Inhibits the Transcriptional Activity of PPARγ...28
6. HBx Inhibits PPARγ-induced Growth Retardation in Hepatoma Cells. ...30
IV. Discussion...33
V. Conclusion ...38
References ...40
List of Figures
Figure 1. Interaction of PPARγ with HBx in vivo. ...20 Figure 2. Interaction of PPARγ with HBx in vitro. ...21 Figure 3. Co-localization of PPARγ with HBx. ...23
Figure 4. Reduced PPARγ protein level in the nucleus of HBx-TG mouse
liver. ...25 Figure 5. Lower PPRE binding of NE from HBx-TG mouse liver tissues. .27 Figure 6. Inhibition of PPARγ transcriptional activity by HBx. ...29 Figure 7. HBx abolishes the inhibitory effect of troglitazone on cellular
proliferation. ...31 Figure 8. HBx abrogates troglitazone-induced cytotoxicity in HepG2 cells. ...32
1
Abstract
Hepatitis B Virus X Protein Modulates Transcriptional Activity of
PPAR
γ
through Direct Protein-Protein Interaction
Youn-Hee Choi
Department of Medical Science The Graduate School, Yonsei University
(Directed by Professor Se Jong Kim)
The X protein of hepatitis B virus (HBx) is a multi-functional protein, which interacts with a number of cellular proteins and plays an important role in the pathogenesis of HBV-associated carcinogenesis. Peroxisomal proliferator-activated receptors (PPARs) are nuclear receptors with pleiotropic effects on metabolism, cell proliferation and oncogenesis. In this study, it was demonstrated that HBx interacted with PPARγ in the cytoplasm and inhibited the transcriptional activity of PPARγ by blocking nuclear translocation. It was also shown that HBx formed complexes with PPARγ in vitro and in vivo. Transient transfection of HBx induced a redistribution of
2
expression of HBx (HBx-TG) in C57BL/6 mice did not alter expression of PPARγ in the liver tissues, while the amount of nuclear PPARγ was significantly reduced in the liver of HBx-TG mice compared to the normal control. Moreover, HBx abrogated the DNA binding of PPARγ. Αddition of recombinant HBx in nuclear extract abrogated the DNA binding activity of PPARγ. HBx suppressed the reporter activity containing PPARγ-response element in a dose-dependent manner. Treatment with troglitazone, a synthetic PPARγ ligand, suppressed proliferation of HepG2 cells, while the same treatment had no inhibitory effect on cell growth of HepG2 cells stably transfected with a plasmid carrying four tandem copies of the HBV genome. Also, troglitazone-induced cytotoxicity in HepG2 cells infected with Ad-GFP-HBx was significantly decreased as compared with that in HepG2 infected with Ad-GFP.
In conclusion, it was demonstrated that HBx interfered with the nuclear translocation of PPARγ by forming complexes with PPARγ in the cytoplasm, leading to abrogation of transcriptional activity and pro-apoptotic properties of PPARγ. Therefore, HBx might play an important role in the pathogenesis of HBV-associated liver diseases by modulating the function of PPARγ.
3
Hepatitis B Virus X Protein Modulates Transcriptional Activity of
PPAR
γ
through Direct Protein-Protein Interaction
<Directed by Professor Se Jong Kim>
Department of Medical Science The Graduate School, Yonsei University
Youn-Hee Choi
I. Introduction
Hepatitis B virus (HBV) is an etiological agent of acute and chronic hepatitis and hepatocellular carcinoma (HCC). Chronic infection of HBV has been known as one of the risk factors in hepatocarcinogenesis1. The 3.5 kb genome of HBV has four open
reading frames: S/preS, C/preC, P and X, which encode surface antigen, core antigen, polymerase and HBx, respectively. Among these, HBx is an essential molecule in viral replication2 and hepatocyte transformation3, 4. However, the molecular mechanism
involved in development of HBV-associated HCC remains largely unknown.
4
functional outcomes, apoptosis and proliferation of liver cells. It has been known that HBx increased both formation of adenoma and apoptosis of hepatocytes in TG mice5.
Furthermore, it is still controversial whether these two events are related to each other or not. It has been proposed that chronic infection of HBV causes both liver damage and compensatory regeneration of hepatocytes at the same time, leading to accumulation of genetic defects and hepatocarcinogenesis6.
HBx can induce aggregation of mitochodria and disruption of transmembrane potential through mitochondrial outer membrane, leading to apoptotic cell death of human hepatoma cell lines7. It has also been reported to sensitize liver cells to tumor
necrosis factor-α (TNFα)-induced apoptosis via activation of mitogen-activated-protein kinase kinase 1 (MEKK1) and accumulation of Myc mitogen-activated-protein8, 9.
Other evidences have been reported that HBx induces cell proliferation and transformation by suppression of p53-induced apoptosis10-12. HBx promotes cellular
transformation, as demonstrated by an immortalization of primary mouse hepatocytes4
and increased occurrence of hepatoma in HBx-transgenic (TG) mice3, 13, 14. HBx can
also activate a variety of signaling pathways such as Ras-Raf-MAP kinase, PI3 kinase, protein kinase C, Src kinase, NF-κB and JAK-STAT pathways, which are responsible for cellular proliferation and carcinogenesis15. In addition, translocation of p53 is
5
HBx has been believed to play important roles as a muitifunctional transactivator, however, it has no known DNA binding motif. Thus, it is likely that HBx mediates its biologic activity through protein-protein interactions rather than direct binding to cis-elements of genomic DNA. Oncogenic activity of HBx is thought to be mediated by interaction with transcriptional factors such as RPB5 of RNA polymerase16, TATA
binding protein17, transcription factor IIB18, bZIP protein19 and retinoid X receptor
(RXR)20.
Peroxisome proliferator-activated receptors (PPARs) belong to the steroid hormone receptor superfamily, and regulate expression of genes associated with differentiation, proliferation and metabolism of fatty acid/glucose21-23. Initially, these receptors are
discovered to be activated by peroxisome proliferators and endogenous fatty acids. Three types of PPARs have been characterized: PPARα, PPARβ (or NUC-1, PPARδ) and PPARγ. Different function and expression of these species have been reported in vivo24. Human PPARγ gene has three isoforms, PPARγ1, PPARγ2 and PPARγ3 by an alternative splicing and differential use of promoters25, 26. Thus, the PPAR family
consists of five isoforms generated from three distinct genes.
Similar to other transcriptional factors, PPARs are composed of three functional domains: a transactivation domain (TAD), DNA binding domain (DBD) and ligand binding domain (LBD). The sequence of TAD is variable between PPAR isoforms,
6
however, the sequence of DBD is largely conserved between different isoforms. DBD possesses two zinc finger motifs, and LBD contains a hydrophobic ligand binding pocket27. A hinge region exists in the N-terminus of LBD and participates in nuclear
localization and formation of heterodimers. Upon receptor ligation, PPARs form heterodimers with the RXR28, 29. These heterodimers bind to specific DNA regions
called PPAR response elements (PPRE) in the 5´ flanking region of target genes28, and
form complexes with coactivators and corepressors involved in PPARs/RXR-mediated transactivation30.
Different PPAR isoforms are widely expressed in variable tissues, however, the concentration of each PPAR varies according to tissue types. For example, PPARα is highly expressed in liver, heart, kidney; PPARβ is expressed ubiquitously in various organs; PPARγ is expressed in higher amount in adipose tissues, liver, kidney and heart, while the expression is lower in lung, testis,brain, skeletal muscle and spleen31, 32. This
distinct tissue expression and different expression level imply that five isoforms of PPARs play different biological roles in different organs and are regulated by a isoform-specific manner.
PPARs mediate pleiotropic functions: PPARα enhances transcription of genes
involved in fatty acid oxidation33 and PPARγ promotes adipocyte differentiation,
7
the role of PPARβ.In addition, PPARs contribute to carcinogenesis and induction of
apoptosis. Wy14643, a ligand of PPARα, has been described as a strong hepatic
mitogen in mice. Treatment with this drug for 11 months resulted in formation of hepatic adenomas and carcinomas in wild type mice but not in PPARα-null mice36, 37.
Whereas, PPARα ligands induce apoptosis in human macrophages, suggesting a cell type-specific effect of PPARα ligands. It has been reported that PPARβ was highly expressed in human primary colorectal carcinomas38. Moreover, induction of APC, a
tumor suppressor gene, causes a decrease of PPARβ expression39, suggesting that
PPARβ is involved in the pathogenesis of human colorectal cancers.
PPARγ ligands induce growth retardation, terminal differentiation and apoptosis in various human cancer cells, including colon cancer, prostate cancer, breast cancer, liposarcoma and hepatoma40-45, thus raising the possibility of an anti-oncogenic and
pro-apoptotic property of PPARγ. It has been suggested that PPARγ induces expression of genes involved in differentiation and cell-cycle46 and suppresses expression of
anti-apoptotic Bcl-247. Therefore, PPARγ ligands such as troglitazone, pioglitazone and
15d-prostaglandin J2 have been proposed as putative chemotherapeutic agents.
However, the molecular mechanisms of PPARγ-mediated apoptosis and cell-cycle
8
In this study, it was demonstrated that HBx formed a complex with PPARγ in vitro and in vivo and suppresses transcriptional activity and apoptosis induced by PPARγ. It was further shown that HBx interruppted the nuclear localization of PPARγ. Therefore, this study proposes a novel association between HBx and PPARγ, which may provide an important mechanism in the pathogenesis of HBV-associated liver diseases.
9
II. Materials and methods
1. Cell lines and culture
The human liver cell lines, Chang liver (ATCC CCL 13) and 293 (ATCC CRL 1573), were maintained in Dulbecco’s Modified Eagles Medium (DMEM) (Gibco BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco BRL).
HepG2 (ATCC HB 8065) and HepG2.2.1548 were maintained in minimal essential
medium (MEM) supplemented with 10% fetal bovine serum. Chang X-34 cells are Chang liver cells expressing HBx under the control of tetracycline inducible promoter10, and were maintained in DMEM with 20 µg/ml of G418 (Gibco BRL). HBx
was induced with 2 µg/ml of doxycycline (Sigma, St. Louis, MO, USA).
2. Plasmids and plasmids construction
A series of GFP-PPARγ and His-PPARγ fusion constructs shown in Fig. 1B and Fig. 2A were respectively generated by PCR amplification of the PPARγ gene with specific primer sets, corresponding to the PPARγ full (F; residues 1-475), PPARγ trans-activation domain (N; residues 1-181), PPARγ DNA binding domain (D; residues
94-181), PPARγ DNA and ligand binding domain (DL; residues 94-475), and PPARγ
ligand binding domain (L; residues 281-475). Bacterial expression and purification of PPARγ proteins were performed as described previously49. To generate His-HBx fusion
10
protein, an amplified HBx fragment was cloned into EcoR I-Hind III sites of pRSET A (Invitrogen, Carlsbad, CA, USA). Minimal PPRE-tyrosine kinase-luciferase reporter construct containing three tandem repeats of the PPAR response element (PPRE3
-tk-LUC) and mouse PPARγ expression plasmid (pCMX-PPARγ)50 were kindly provided
by Dr. R. Evans. HBx DNA construct under the control of SV40 promoter (pSVX) and
its frameshift mutant (pSVXκB)51 were used for transfection experiments. To
determine intracellular interaction of PPARγ with HBx and subcellular localization, PPARγ expression plasmids (pEGFP-PPARγ F, N, D, DL, L) were constructed by PCR amplification and subcloned into the Sac I-Sal I site of pEGFP vector (Clontech, Palo Alto, CA, USA). To express HBx with hemagglutinin (HA) epitope tag at the N terminal (pCMV-HA-HBx), the HBx fragment was amplified by PCR and subcloned into the EcoR I–Kpn I site of pCMV-HA plasmid (Invitrogen).
3. HBx-TG mice
HBx-TG mice were generated and maintained as described previously14. Animals
have been maintained as an inbred strain by sister-brother mating under SPF conditions. Age matched wild type control mice were used as controls. All mice were caged in groups of four in an animal room with constant temperature (25±1°C) and humidity (60±5°C) and a 12-hour light cycle. Rats were fed a normal diet (Shinchon Diet Co., Korea) containing 20% protein. the procedure of our experiment was approved by
11
IACUC (Institutional Animal Care and Use Committee) in Yonsei University according to the “Guideline for Animal Experiments” revised by Korean Academy of Medical Sciences (2000).
4. Antibodies and reagents
Anti-PPARγ antibodies (PA3-821, Affinity Bioreagents, Golden, CO; sc-7273, Santa Cruz Biotechnology, Santa Cruz, CA, USA), goat anti-SP-1 polyclonal antibody (sc-59-G, Santa Cruz Biotechnology), mouse anti-α-tubulin monoclonal antibody (CP-06, Oncogene, Darmstadt, Germany), rabbit anti-HA polyclonal antibody (sc-805, Santa Cruz Biotechnology) and mouse anti-GFP monoclonal antibody (sc-805, Santa Cruz Biotechnology) were used for western blot analysis. Immunoprecipitation was performed using an anti-HA monoclonal antibody (sc-7392, Santa Cruz Biotechnology). Troglitazone, provided by SanKyo (Tokyo, Japan), was prepared as previously described52. Oxidized and reduced glutathione (GSH, GSSH), dithiothreitol
(DTT) and isopropyl β-D-thiogalatopyranoside (IPTG) were purchased from Sigma. Nickel agarose beads were supplied by Qiagen (Qiagen Inc., Chatsworth, CA, USA).
5. Immunoprecipitation and Western blot analysis
12
treated with 2 µg/ml of doxycycline for 4, 9, and 24 h, and then lysed with RIPA buffer containing 50 mM Tris-Cl, pH 7.4, 100 mM NaCl, 0.5 mM EDTA, 0.2% NP-40 and 2 mM MgCl2 supplemented with protease inhibitor cocktail (Sigma). For detection of
intracellular interaction of HBx with deletion mutants of PPARγ, 293 cells were co-transfected with HA-HBx and GFP-PPARγ F, N, D, DL, or L, respectively. Then, cell lysates (500 µg) were incubated with 1 µg of anti-HA monoclonal antibody and incubated with Protein G/A agarose (Roche Molecular Biochemicals, Mannheim, Germany) for 18 h. Western blot analyses were performed using polyclonal anti-HA,
anti-PPARγ antibody and anti-GFP antibody. Goat anti-rabbit antibody (DAKO,
Glostrup, Denmark) conjugated with horseradish peroxidase (HRP) and HRP-conjugated goat anti-mouse antibody (Amersham Phamacia, Uppsala, Sweden) were used as the secondary antibodies. Reactive proteins were detected by incubating in SuperSignalTM substrate (Pierce, Rockford, IL, USA) and exposing to X-ray film
(Eastman Kodak Company, Rochester, NY, USA).
6. In vitro binding assay and native gel shift assay
His fusion constructs, His-PPARγ D, DL, L were transformed into E. coli strain BL21 (DE3). His fusion proteins were purified by affinity chromatography using Nickel-Agarose 4B bead (Qiagen Inc.) and further purified on an FPLC gel-filtration column. Protein expression, preparation of inclusion body proteins, refolding of
13
denatured proteins and native gel shift assay were performed as described previously53.
The recombinant soluble proteins were mixed in equal molarities and incubated overnight at 4ºC. The reaction mixtures were then assayed by 12% non-denaturing polyacrylamide gel electrophoresis.
7. Immunofluorescent assay (IFA)
IFA was used to investigate subcellular localization of HBx and PPARγ. Briefly, Chang liver cells were seeded into Lab-Tek two chambers glass slides (Nalge Nunc, Naperville, IL, USA) and transiently transfected with pEGFP-PPARγ and pCMV-HA-HBx. Cells were incubated in the absence or presence of 20 µM troglitazone for 24 h, washed and fixed with 4% paraformaldehyde in PBS and permeabilized with 0.1% saponin (Sigma) in PBS. Cells were incubated with a rabbit anti-HA polyclonal antibody then with Cy3-conjugated goat anti-rabbit IgG (Zymed Laboratories Inc., South San Francisco, CA, USA). After several washes, cells were mounted using VECTASHIELD mounting medium containing DAPI (Vector Laboratories Inc., Burlingame, CA, USA) and visualized under a confocal microscope (Carl Zeiss, Jena, Germany).
14
8. Northern blot analysis
Total RNA was prepared using a Qiagen RNeasy kit (Qiagen Inc.) and Northern blot analysis was performed as described previously with a minor modification54. Fifteen
micrograms of total RNA obtained from the livers of control C57BL/6 and HBx-TG mice was electrophoresed in 1% agarose gel containing 6% formaldehyde/1X MOPS and transferred to Hybond-N nitrocellulose membrane (Amersham Pharmacia). Probe labeling was performed using a random primer labeling kit (Promega, Madison, WI, USA). Membrane was hybridized with 32P-labeled PPARγ or β-actin probes using
ExpressHybTM (Clontech). The blot was washed and visualized by autoradiography.
9. Electromobility shift assay (EMSA)
Preparation of nuclear extract (NE) and EMSA were carried out as previously
described52. Liver tissues were homogenized by a Dounce homogenizer and
resuspended in 1 ml of lysis buffer containing 10 mM Tris-Cl, pH 7.4, 3 mM CaCl2,
and 2 mM MgCl2, and then centrifuged at 1,500 rpm for 15 min. Supernatant was
removed, and cell pellet was resuspended in 1 ml of lysis buffer containing 10 mM Tris-Cl, pH 7.4, 3 mM CaCl2, 2 mM MgCl2, and 1% NP-40. Nuclei were harvested by
centifugation and nuclear proteins were further extracted with buffer C containing 20
15
EDTA, 0.5 mM DTT, and 0.5 mM PMSF for 40 min in cold room using an orbital shaker. NEs were obtained after centrifugation at 14,000 rpm for 30 min. For binding reaction, 5 µg of NE was incubated with the probe in the binding buffer containing 10 mM Tris-Cl, pH 7.5, 100 mM KCl, 1 mM DTT, 1 mM EDTA, 0.2 mM PMSF, 1 mg/ml BSA, and 5% glycerol at RT for 30 min. For the competition experiments, a 100 molar excess of unlabeled probe28 was added to the reaction mixture. To examine the effect of
HBx on DNA binding activity of PPARγ, recombinant HBx was added to reaction
mixture for 30 min, then probes were added. The reaction mixtures were analyzed in a 5% polyacrylamide gel and electrophoresis was carried out at 150V for 3 h. After drying, the gel was visualized by autoradiography. For SP-1 binding, 2 µg of NE was incubated with the labeled probe. The sequence of the oligonucleotides used as probes was as follows: CYP4A-PPRE, TGAAACTAGGGTAAAGTTCA-3´; SP-1, 5´-GATCGATCGGGGCGGGGCGAG-3´.
10. Luciferase reporter assay
Chang liver cells (2 x 105) were seeded onto 12 well tissue culture plates and
transfection was carried out using LipofectAMINE PLUSTM reagent (Gibco BRL)
according to the manufacturer’s instruction. The total amount of DNA was adjusted to
1 µg; 500 ng of PPRE3-tk-LUC, 200 ng of pCMX-PPARγ, 200 ng of pSVX or
16
the cells were washed and incubated in a complete medium in the absence or presence of 20 µM troglitazone for 24 h. Cells were then washed and lysed using 5 x lysis buffer (Promega). Lysates were measured for luciferase activity using luciferin (Promega), and β-galactosidase activity was measured as an internal control.
11. Cytotoxicity assay
Cell growth was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay55.Briefly, 1 x 103 cells were seeded to 96-well plates and treated
with troglitazone for different time periods. Cells were then washed in PBS, pH 7.4, added 50 µl of MTT solution (1 mg/ml in PBS), and incubated for 4 h at 37 ℃. The plate was then centrifugated at 3,000 rpm for 10 min, supernatant was carefully
removed, and remaining adhered cells were dissolved in 50 µl of DMSO (Sigma).
Optical density of each well was measured at 570 nm using ELISA reader.
12. Generation and infection of recombinant adenoviruses
Construction of adenoviral vectors was performed as described previously56. The
adenoviral plasmids (pAdEasy-1 and pAdEasy-2) and the shuttle vectors (pShuttle, pShuttle-CMV, pAdTrack, and pAdTrack-CMV) were constructed through subcloning of PCR product of HBx. PCR-derived HBx fragments were sequenced to confirm their predicted composition. Approximately 3 x 106 293 cells were plated in 25 cm2 flasks
17
24 hr before transfection, by which time they reached 50–70% confluency. Four micrograms of recombinant adenoviral vector DNA, digested with PacI and ethanol precipitated, were used for transfection. A transfection mix was prepared by adding 4 µg of linearized plasmid DNA and 20 µl of LipofectAMINE (Gibco BRL) in 293 cells. Transfected cells were monitored for GFP expression and harvested 7 days after transfection. Cells were collected and lysed by three cycles of freezing/thawing. Concentrated adenovirus fraction was prepared by CsCl gradient centrifugation and stored with 2X storage buffer (10 mM Tris, pH 8.0, 100 mM NaCl, 0.1% BSA, and 50% glycerol, filter sterilized) at -20°C. One mililiter of viral lysate was used to infect 3–5 x 106 cells. The efficiency of infections was monitored by GFP expression.
18
III. Results
1. HBx Binds to PPAR
γ
in vivo and in vitro.
To examine whether HBx can bind to PPARγ, an immunoprecipitation assay was
performed with Chang X-34 cell lines, which express HBx-HA under the control of the tetracycline inducible promoter. Chang X-34 cells were incubated in the presence of
doxycycline (2 µg/ml) for various time periods. Cell lystes were prepared,
immunoprecipitated with an anti-HA monoclonal antibody, and then analyzed with an anti-PPARγ antibody. The HBx-PPARγ complex was increased in proportion to the
HBx expressed by doxycycline in a time-dependent manner, although the PPARγ
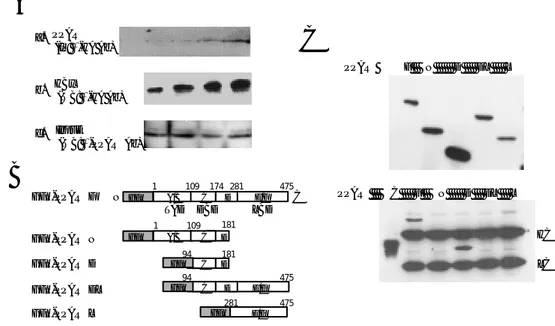
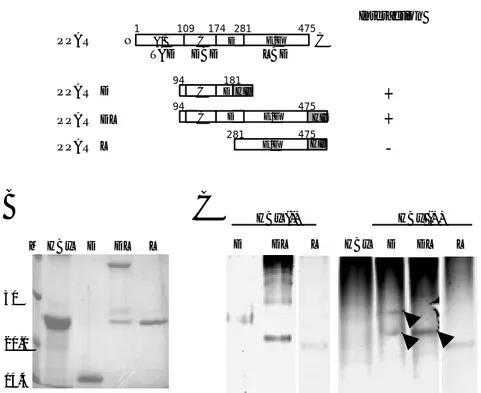
protein level was unaffected by HBx expression (Fig. 1A). To verify which domain of PPARγ isinvolved in the binding with HBx, 293 cells were transfected with HA-HBx and a series of GFP-PPARγ fusion constructs (Fig. 1B). As shown in Fig. 1C, PPARγ F and D was associated with HBx. These results demonstrate the physical interaction of HBx with PPARγ.
Since it has been reported that RXRα can bind to HBx and heterodimerize with
PPARγ29, an in vitro binding assay using recombinant His-HBx and His-PPARγ
proteins was performed to exclude the possibility that HBx binds to PPARγ indirectly thorough RXRα. HBx and three PPARγ constructs were generated as shown in Fig. 2A.
19
Refolded proteins were purified by gel filtration chromatography, and peak fractions were analyzed by SDS-PAGE (Fig. 2B). To verify which domain of PPARγ isinvolved in the binding with HBx, recombinant HBx protein was mixed with three His-PPARγ fusion proteins in PBS for 18 h, and a native gel shift assay was performed. Native gel shift assay showed newly formed bands or shifted bands in the mixtures of refolded HBx, and PPARγ D and PPARγ DL proteins (Fig. 2C, shown as arrow heads), but not in the mixtures of refolded HBx and PPARγ L. It was found that PPARγ D and PPARγ DL proteins interacted with HBx through the DNA binding domain and the hinge region of PPARγ. These results indicate that HBx binds directly to PPARγ via its DNA binding domain and the hinge region, which have an important role in the nuclear translocation and DNA binding of PPARγ.
20
HBx
(WB: α-HA Ab)
PPARγ
(IP: α-HA Ab)
Input (WB: α-PPARγAb) a. b. c.
A
B
GFP-PPARγF GFP-PPARγL GFP-PPARγDL GFP-PPARγD GFP E/F A/B C D E/F TAD DBD LBD N C 1 109 174 281 475 D E/F 94 475 C D 94 181 281 475 C A/B C 1 109 181 D GFP GFP GFP GFP GFP-PPARγNC
PPARγ F N D DL L PPARγ C F N D DL L HC LCFigure 1. Interaction of PPARγ with HBx in vivo. (A) After treating Chang X-34 cells with 2 µg/ml doxycycline, (a) cell lysate was immunoprecipitated with anti-HA monoclonal antibody and analyzed on 10% SDS-PAGE gel. Western blot was performed with anti-PPARγ antibody. C, immunoprecipitation with normal mouse serum; (b) Western blot with anti-HA antibody; (c) Western blot with anti-PPARγ, showing that an equal amount of PPARγ had been loaded. (B) A schematic diagram of PPARγ and four GFP-PPARγ fusion constructs encoding the indicated domain. (C) 293 cell lysates were analyzed by Western blot with anti-GFP antibody showing that HBx and PPARγ fusion proteins were well expressed. After transfection with HA-HBx and GFP-PPARγ fusion constructs, 293 cells were lysed and cell lysates were immunoprecipitated with anti-HA monoclonal antibody. Western blot was performed with mouse anti-GFP monoclonal antibody. C, immunoprecipitation with normal mouse serum; HC, heavy chain; LC, light chain.
21
B
M HBx 30 20.1 14.4 D DL LC
D DL HBx (-) L HBx D DL HBx (+) LA
PPARγ PPARγL PPARγDL PPARγD HIs E/F A/B C D E/F TAD DBD LBD N C 1 109 174 281 475 D E/F 94 475 HIs C D 94 181 281 475 HIs C Interaction + +-Figure 2. Interaction of PPARγ with HBx in vitro. (A) A schematic diagram of
His-PPARγ fusion constructs encoding the indicated domain. (B) SDS-PAGE of the
purified and refolded His-fusion proteins. M: marker (C) Native gel shift assay revealed that HBx binds to PPARγ fusion proteins containing DBD and the hinge region.
22
2. HBx Alters the Subcellular Localization of PPAR
γ
.
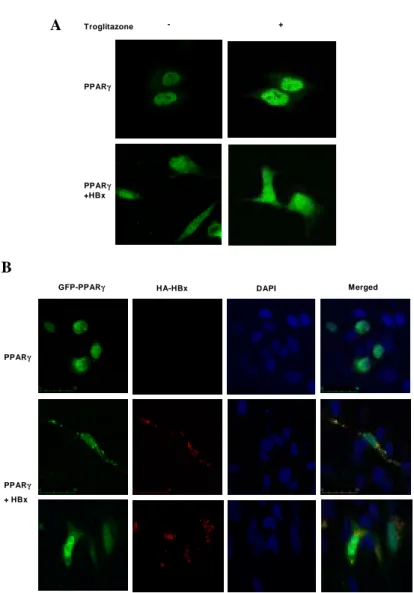
To examine whether this binding affects the nuclear localization of PPARγ, IFA was
performed after co-transfecting pEGFP-PPARγ and pCMV-HA-HBx into Chang liver
cells, and analyzing the distribution of GFP-PPARγ. In this experiment, PPARγ was found to be located mainly in the nucleus in the presence or absence of troglitazone (Fig. 3A, upper panel). Interestingly, a large amount of GFP-PPARγ was detected in the cytoplasm under HBx overexpressing conditions in spite of troglitazone treatment, although some amount of PPARγ was still present in the nucleus (Fig. 3A, lower panel).
Since much more PPARγ was present in the cytoplasm under conditions of HBx
overexpression, it was determined whether HBx and PPARγ colocalize in the
cytoplasm, as in the case of p5310-12. As shown in Fig. 3B, cytoplasmic PPARγ was
found to be co-localized with HBx (merged; yellow color). This result indicates that HBx interferes with the nuclear localization of PPARγ by complex formation in the cytoplasm.
23 PPARγ Troglitazone PPARγ +HBx + -A B PPARγ PPARγ + HBx
GFP-PPARγ HA-HBx DAPI Merged
Figure 3. Co-localization of PPARγ with HBx. (A) pEGFP-PPARγ with or without pSVX-HBx was transfected into Chang liver cells and treated with or without 20 µM troglitazone, cells were observed under a confocal microscope. Partial cytoplasmic sequestration of PPARγ was observed in the presence of HBx. Upper panel, GFP-PPARγ transfected Chang liver cells; lower panel, GFP-PPARγ and HBx co-transfected Chang liver cells. (B) pEGFP-PPARγ was transfected into Chang liver cells with or without pCMV-HA-HBx. After incubation with 20 µM troglitazone for 20 hrs, the cells were stained and then analyzed under a confocal microscope. Cells were visualized with triple colors, GFP (PPARγ), and rabbit anti-HA antibody and Cy3 conjugated anti-rabbit IgG (HBx), and DAPI(nucleus), and merged.
24
3. Expression of PPAR
γ
in the HBx-TG Mouse Liver Is Reduced in the
Nucleus.
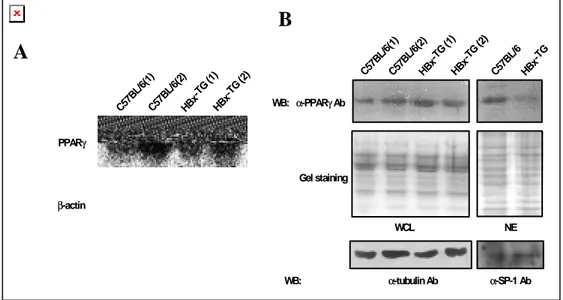
To investigate whether the expression of HBx influences the PPARγ expression, the mRNA expression of PPARγ was determined by Northern blot in HBx-TG and control C57BL/6 mouse liver tissues. In agreement with a previous report31, PPARγ mRNA
was expressed in the liver tissue (Fig. 4A). Western blot analysis was used to compare PPARγ protein expression levels in the whole cell lysate (WCL) and in the nuclear extreact (NE) preparation of the control C57BL/6 and HBx-TG mouse liver tissues. No significant difference was found in the protein expression levels in the WCL of
HBx-TG or in the control C57BL/6 mouse liver tissues. Interestingly, however, the
expression of PPARγ in the NE preparation of the HBx-TG mice was significantly lower than in that of the control mice (Fig. 4B).
25 WB: α-PPARγ Ab C57 BL/6( 1) C57 BL/ 6(2) HBx -TG (1) HBx -TG (2) HBx -TG C57 BL/ 6 Gel staining WCL NE
B
WB: α-tubulinAb α-SP-1 Ab PPARγ β-actinA
C57 BL/ 6(1) C57 BL/6( 2) HBx -TG (1) HBx -TG (2)Figure 4. Reduced PPARγ protein level in the nucleus of HBx-TG mouse liver. (A) Northern blot showing the mRNA expression of PPARγ in HBx-TG mouse liver and in the control C57BL/6. (B) PPARγ protein expression. WCL and NE preparations were obtained, as described in Materials and Methods. Fifty micrograms of WCL or 15 µg NE was analyzed on a 10% SDS-polyacrylamide gel and Western blot was performed using anti-PPARγ Ab. Coomassie brilliant blue R-250 staining and Western blot with anti-α-tubulin and anti-SP-1 antibody were used to confirm equivalent gel loading.
26
4. The DNA Binding of PPAR
γ
Is Reduced in HBx-TG Mouse.
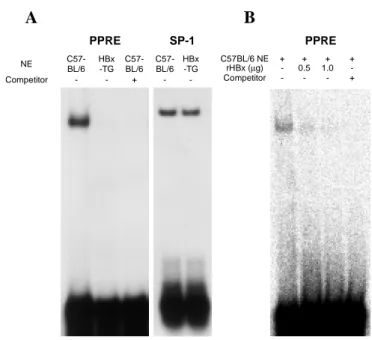
Since HBx interacts directly with PPARγ, it was examined whether this binding influences the DNA binding activity of PPARγ. To test this hypothesis, EMSA was used with PPRE to determine the effect of HBx on the DNA binding of PPARγ. NE was prepared from the liver of 13 months-old HBx-TG mice and age controlled C57BL/6 mice. As shown in Fig. 5A, the NE from the control C57BL/6 mouse liver showed DNA binding to the PPRE sequence, however, the DNA binding of PPARγ was completely abrogated in the NE of HBx-TG mouse liver. To confirm that HBx inhibits
the DNA binding of PPARγ, we performed EMSA using NE from control C57BL/6
mouse liver which was pre-incubated with recombinant HBx protein. Fig. 5B shows that the recombinant HBx protein significantly reduced the DNA binding activity of PPARγ.
27 - C57-BL/6 -HBx -TG + -Competitor C57-BL/6 HBx -TG C57-BL/6 NE PPRE SP-1 A B -1.0 0.5 -rHBx (µg) -+ + + -Competitor + + C57BL/6 NE PPRE
Figure 5. Lower PPRE binding of NE from HBx-TG mouse liver tissues. (A) Five micrograms of NE from control C57BL/6 and HBx-TG mouse liver was analyzed by EMSA, as described in Materials and Methods. For the competition assay, a 100 molar excess of the cold probe was added to the reaction mixture. The binding of SP-1 was shown as a control. (B) NE from control C57BL/6 mouse liver was pre-incubated at RT for 30 min with or without the indicated concentration of recombinant HBx protein before addition of the probe.
28
5. HBx Inhibits the Transcriptional Activity of PPAR
γ
.
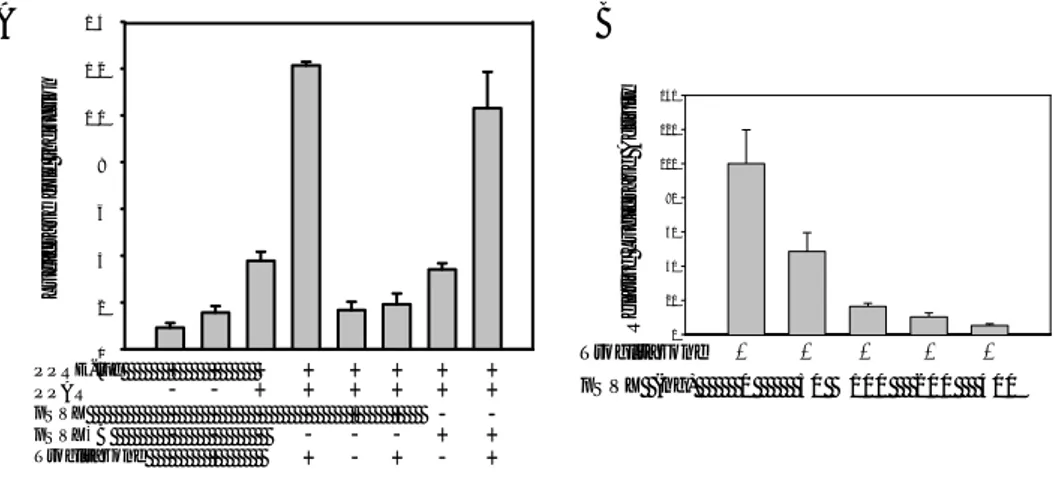
To examine whether HBx modulates the transcriptional transactivation activity of PPARγ, luciferase reporter assays were performed using PPRE3-tk-LUC. Compared
with Chang liver cells transfected with PPRE3-tk-LUC alone, luciferase activity was
increased by PPARγ overexpression (a 4 fold induction versus the empty vector) and this activity was enhanced by troglitazone (a 13 fold induction versus the empty vector). However, this increase in the transcriptional activity of PPARγ was markedly abrogated by HBx expression even in the presence of troglitazone, but not by mutated HBx (Fig. 6A), and this inhibition of PPARγ transcription activity by HBx occurred in a dose-dependent manner (Fig. 6B). Therefore, these results strongly suggest that HBx inhibits the transcriptional activity of PPARγ.
29 0 2 4 6 8 1 0 1 2 1 4 A PPRE-luc PPARγ pSVX pSVXκB T roglitazone - + - + - + - + + + -- - - - + + - -+ + + + + + -- + + + + + + + + Lu ci fer a s e fol d i nduc ti on B 0 20 40 60 80 100 120 140 pS V X (ng) 0 50 100 20 0 400 T roglitazone + + + + + R e la ti v e Luc if er a s e Acti vit y
Figure 6. Inhibition of PPARγ transcriptional activity by HBx. Transient
transfection in Chang liver cells and the luciferase reporter assay were used to examine the effect of HBx on the PPARγ transcriptional activity, as described in
Materials and Methods. (A) PPRE3-tk-LUC activity was inhibited by the
overexpression of HBx but not by mutated HBx. Each bar was normalized versus β -galactosidase activity. Luciferase activities are represented as fold induction relative to the basal activity of the empty vector in the absence of troglitazone. (B) Dose-dependent effect of HBx on PPRE3-tk-LUC activity. Normalized values are shown as
means ± SD of independent experiments performed in triplicate and repeated at least 3 times.
30
6. HBx Inhibits PPAR
γ
-induced Growth Retardation in Hepatoma Cells.
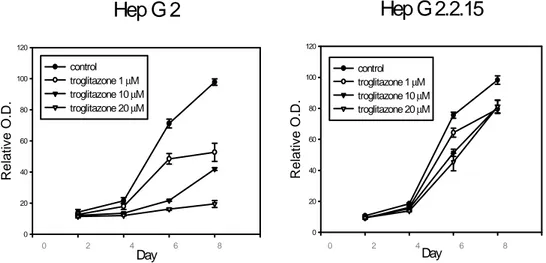
In order to examine the physiologic relevance of the inhibitory effect of HBx on PPARγ, MTT assay was performed with HepG2 and HepG2.2.15 cells after treatment of a PPARγ ligand, troglitazone. HepG2.2.15 cell line was established by transfecting with a plasmid carrying four tandem copies of the HBV genome into HepG2 cell48. As
mentioned above, PPARγ ligands reveal anti-proliferative effect on the various cancer cell lines including hepatoma40-45. To examine whether HBx represses the growth
inhibitory effect of PPARγ ligand on hepatoma cells by inhibition of PPARγ function, proliferation of HepG2, HepG2.2.15 and HepG2 cells infected with recombinant adenovirus carrying HBx was measured for the different time periods after treatment with troglitazone. When the cells were treated with troglitazone the proliferation of HepG2 cells significantly decreased in a dose-dependent manner (Fig. 7). However, HepG2.2.15 cells appeared to be more resistant to troglitazone-induced growth inhibition than HepG2. In an attempt to clarify the inhibitory effect of HBx on troglitazone-induced cytotoxicity, adenoviruses carrying GFP-HBx or GFP genes were administered into HepG2 cells, and cytotoxicity was measured for the different time periods after treatment with 20 µM troglitazone. As shown in Fig. 8, survival in HepG2 cells infected with Ad-GFP-HBx was significantly increased as compared with that in HepG2 infected with Ad-GFP. Thus, these results demonstrated that HBx significantly repressed the pro-apoptotic function of PPARγ.
31
Hep G 2.2.15
Day Re la ti v e O .D. 0 20 40 60 80 100 120 control troglitazone 1 µM troglitazone 10 µM troglitazone 20 µMHep G 2
Day Re lat iv e O .D . 0 20 40 60 80 100 120 control troglitazone 1 µM troglitazone 10 µM troglitazone 20 µM 0 2 4 6 8 0 2 4 6 8Figure 7. HBx abolishes the inhibitory effect of troglitazone on cellular proliferation. HepG2 and HepG2.2.15 cells were treated with 1.0 µM to 20 µΜ troglitazone for the indicated time periods and then cellular proliferation was determined by MTT assays. Results are represented as relative O.D. compared with control. Data was shown the mean ± SD of four independent experiments.
32
Day
0 1 2 3 4 5 6 7Re
la
ti
v
e
O.D
.
0 1 2 3 4 5 HepG2-AdGFP HepG2-AdGFP-HBxFigure 8. HBx abrogates troglitazone-induced cytotoxicity in HepG2 cells. HepG2 cells were infected with recombinant adenovirus carrying HBx (Ad-GFP-HBx) or control virus (Ad-GFP) as described in Materials and methods. Cells were treated with 20 µΜ troglitazone for the indicated time periods and then cellular proliferation was determined by MTT assays. Results are represented as relative O.D. compared with control. Data was shown the mean ± SD of four independent experiments.
33
IV. Discussion
HBx is known to have a transcriptional transactivation property, mediating various functions through protein-protein interactions with endogenous cellular proteins and transcriptional factors. In the present study, viral protein HBx directly interacts with the DNA binding domain including the hinge region of PPARγ. In addition, it is shown that HBx suppresses the transcriptional activity of PPARγ, by blocking the nuclear localization and DNA binding of PPARγ.
PPARγ is a well-known nuclear hormone receptor with diverse functions, some of which are critical for maintaining cellular processes57. Activation and regulation of
PPARγ occur through the presence of specific ligands and heterodimerization with RXR29. PPARγ also needs other proteins, like co-activators or co-repressors, to
facilitate its full activation, heterodimerizing, binding to chromatin, initiating and activating of basal transcriptional machinery, and promoting downstream gene expression via protein-protein interaction30. PPARγ is composed of a central DBD,
nuclear localization signals (NLSs), a ligand binding domain, and a transactivation domain58. Moreover, specific ligand binding at the LBD of PPARγ results in a
conformational change that allows PPARγ to heterodimerize with RXRα, localize in the nucleus, and regulate the transcription of genes containing PPRE sequences. DBD has two zinc-finger motifs that can bind directly to DNA, and the hinge region
34
contains NLS (a.a. 172-185).
In vitro binding assay and immunoprecipitation demonstrated the direct protein-protein interaction between HBx and PPARγ DBD both in vitro and in vivo. DBD is most highly conserved region between other members of steroid hormone receptor
superfamily. This fact leads to speculation that HBx may interact other PPARγ
isoforms, such as PPARα andPPARδ, or other steroid receptor members. Previously, the DBD of several steroid hormone receptors have been reported to interact with DBD-associating proteins. Thioredoxin, one of DBD-associating proteins, interacts with the DBD of glucocorticoid receptor59. A small nuclear ring finger protein SNURF
directly binds to the DBD of androgen receptor60. In addition to these cellular proteins,
viral protein can modulate steroid hormone receptors through direct protein-protein interaction. Hepatitis C virus core protein interacts with the DBD of RXRα61. Indeed,
coactivators and corepressors for steroid receptors have been reported to bind to the LBD or TAD of steroid receptors62, 63. Adenovirus E1A interacts with the LBD of
RARβ64. The Epstein-Barr virus BZLF-1 was also shown to interact with the LBD of
RXRα and RARα, and to repress the transcriptional activity of RXRα65. As
mentioned above, it was reported that HBx can bind to the LBD of RXR and augment RXR function20.
35
localization of PPARγ, inhibit DNA binding, and significantly suppress the
transcriptional transactivation activity of a reporter gene containing PPAR-response elements. It was speculated that the HBx could modulate the DNA binding of PPARγ, because DBD of PPARγ participated in the interaction with HBx. As expected, HBx
showed the strong inhibitory effect on the PPRE binding of PPARγ in HBx-TG
mice. Moreover, recombinant HBx inhibited the DNA binding activity of PPARγ in
vitro. Interestingly,HBx reduced the amount of nuclear PPARγ in vivo. It suggested a
possibility that another inhibitory mechanisms of HBx remained in addition to
inhibition of DNA binding of PPARγ. IFA data demonstrated that HBx partially
sequestered and colocalized with PPARγ in the cytoplasm. The reason for these results may be explained that HBx affect the DBD and NLS located at the hinge region, leading to the reduction of DNA binding activity and the interruption of nuclear localization of PPARγ. These results suggested that HBx modulated the function of PPARγ by both cytoplasmic sequestration and inhibition of DNA binding to PPRE. Consequently, HBx sufficiently inhibited transcriptional activity and pro-apoptotic property of PPARγ. HBx exists in both cytoplasm and nucleus66, suggesting that HBx
could play an inhibitory role on cytoplasmic and nuclear PPARγ. In other words, partial cytoplasmic sequestration of PPARγ by HBx may sufficiently reduce the concentration of nuclear PPARγ and nuclear PPARγ can not bind to DNA effectively in the presence of HBx, even though some PPARγ exists in the nucleus.
36
Similar to this phenomenon, it has been reported that HBx interferes with p53 function by directly binding and sequestering p53 in the cytoplasm, leading to abrogating p53-mediated cellular processes10-12. Wang et al.67 reported that HBx
inhibits p53 sequence specific DNA binding and blocks transcriptional transactivation by p53 of a reporter gene containing multiple p53-responsive elements. In addition, HBx sequesters p53 in the cytoplasm, leading to abrogating p53-mediated transcriptional transactivation10-12.
Data are accumulating to indicate that PPARγ may induce apoptosis in cancer cells by its ligands. The ligands of PPARγ were reported to induce growth retardation in human cancer cells45, 68, showing the possibility of the anti-cancer and pro-apoptotic
functions of PPARγ. Also, PPARγ ligands have been suggested as putative anti-cancer agents, because of their abilities to induce apoptotic or non-apoptotic cell death69, 70.
However, little is known about the mechanism of PPARγ-mediated gene regulation in apoptosis. In the present experiments, cytotoxicity of troglitazone was observed in HepG2 cells but not in HepG2.2.15 cells, in which stably transfected with a plasmid carrying four tandem copies of the HBV genome in HepG2 cells48.Consistent with this
result, HepG2 cells infected recombinant adenovirus carrying HBx showed more resistance to treatment of troglitazone. HBx suppressed the transcriptional activity of PPARγ and blocked ligand-induced apoptosis significantly. It led to be a speculation that another physiologic role of HBx in modulation of cellular proliferation. Thus,
37
inhibition of PPARγ activity by HBx may induce accelerated cellular proliferation and survival process during HBV infection, leading to chronic hepatocyte proliferation and hepatocarinogenesis. In addition, presumably, it is conceivable that this inhibitory effect of HBx on PPARγ may provide a clonal selective advantage for hepatocytes expressing HBx. HBV is not a cytopathic virus, but leads to liver diseases induced by immune response against its viral antigen. Thus, chronicity is important mechanism allowing it to evade the host immune system. Therefore, these results suggest the possibility that viral protein HBx may play a role in biological modulation in HBV-associated diseases through the interaction with and suppression of PPARγ.
In the present investigation, a novel interaction between HBx and PPARγ was studied. The results obtained from this study showed that HBx inhibited the transcriptional and pro-apoptotic activities of PPARγ. Recognizing the importance of PPARγ-mediated cellular events, and considering the numerous biological properties of HBx, these results imply that this interaction between HBx and PPARγ may play a critical role in the pathogenesis of HBV-associated liver diseases and contribute to the HBV-associated hepatocarcinogenesis.
38
V. Conclusion
This study demonstrates that HBx binds to PPARγ in the cytoplasm and interferes nuclear translocation of PPARγ, to cause suppression of the transcriptional activity and pro-apoptotic property of PPARγ.
1. ΗBx directly interacts with PPARγ in vitro and in vivo.
2. The subcellular localization of PPARγ was redistributed to the cytoplasm by HBx.
3. Transgenic expression of HBx (HBx-TG) in C57BL/6 mice did not alter expression of PPARγ in the liver tissues, while the amount of nuclear PPARγ was significantly reduced in the liver of HBx-TG mice compared to the normal control.
4. HBx inhibits the DNA binding activity of PPARγ.
5. The transcriptional activity of PPARγ was dose-dependently inhibited by HBx.
6. Cell viability was decreased with PPARγ ligand in HepG2, but this effect was not observed in HepG2.2.15.
39
7. Survival in HepG2 cells infected with Ad-GFP-HBx was significantly increased as compared with that in HepG2 infected with Ad-GFP after treatment with troglitazone.
HBx prevents the translocation to the nucleus and binding of PPARγ to DNA due to the complex formation in the cytoplasm, thus inhibits the transcriptional activity of PPARγ. It suggests that HBx modulates the function of PPARγ via protein-protein interaction and this modulation may play a role in the pathogenesis of HBV-associated liver diseases and contribute to the HBV-associated hepatocarcinogenesis.
40
References
1. Feitelson MA. Hepatitis B virus in hepatocarcinogenesis. J Cell Physiol 1999;181:188-202.
2. Nakatake H, Chisaka O, Yamamoto S, Matsubara K, Koshy R. Effect of X protein on transactivation of hepatitis B virus promoters and on viral replication. Virology 1993;195:305-14.
3. Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991;351:317-20.
4. Seifer M, Gerlich WH. Increased growth of permanent mouse fibroblasts in soft agar after transfection with hepatitis B virus DNA. Arch Virol 1992;126:119-28.
5. Koike K, Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Tsutsumi T, et al. Compensatory apoptosis in preneoplastic liver of a transgenic mouse model for viral hepatocarcinogenesis. Cancer Lett 1998;134:181-6.
6. Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, et al. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 1989;59:1145-56.
7. Takada S, Shirakata Y, Kaneniwa N, Koike K. Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene 1999;18:6965-73.
41
killing by tumor necrosis factor alpha. Proc Natl Acad Sci U S A 1997;94:8744-9.
9. Su F, Theodosis CN, Schneider RJ. Role of NF-kappaB and myc proteins in apoptosis induced by hepatitis B virus HBx protein. J Virol 2001;75:215-25.
10. Yun C, Lee JH, Park H, Jin YM, Park S, Park K, et al. Chemotherapeutic drug, adriamycin, restores the function of p53 protein in hepatitis B virus X (HBx) protein-expressing liver cells. Oncogene 2000;19:5163-72.
11. Truant R, Antunovic J, Greenblatt J, Prives C, Cromlish JA. Direct interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx of p53 response element-directed transactivation. J Virol 1995;69:1851-9.
12. Elmore LW, Hancock AR, Chang SF, Wang XW, Chang S, Callahan CP, et al. Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc Natl Acad Sci U S A 1997;94:14707-12.
13. Koike K, Moriya K, Iino S, Yotsuyanagi H, Endo Y, Miyamura T, et al. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology 1994;19:810-9.
14. Yu DY, Moon HB, Son JK, Jeong S, Yu SL, Yoon H, et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J Hepatol 1999;31:123-32.
15. Diao J, Garces R, Richardson CD. X protein of hepatitis B virus modulates cytokine and growth factor related signal transduction pathways during the course of viral infections and hepatocarcinogenesis. Cytokine Growth Factor Rev 2001;12:189-205.
42
16. Cheong JH, Yi M, Lin Y, Murakami S. Human RPB5, a subunit shared by eukaryotic nuclear RNA polymerases, binds human hepatitis B virus X protein and may play a role in X transactivation. EMBO J 1995;14:143-50.
17. Qadri I, Maguire HF, Siddiqui A. Hepatitis B virus transactivator protein X interacts with the TATA- binding protein. Proc Natl Acad Sci U S A 1995;92:1003-7.
18. Haviv I, Shamay M, Doitsh G, Shaul Y. Hepatitis B virus pX targets TFIIB in transcription coactivation. Mol Cell Biol 1998;18:1562-9.
19. Perini G, Oetjen E, Green MR. The hepatitis B pX protein promotes dimerization and DNA binding of cellular basic region/leucine zipper proteins by targeting the conserved basic region. J Biol Chem 1999;274:13970-7.
20. Kong HJ, Hong SH, Lee MY, Kim HD, Lee JW, Cheong J. Direct binding of hepatitis B virus X protein and retinoid X receptor contributes to phosphoenolpyruvate carboxykinase gene transactivation. FEBS Lett 2000;483:114-8.
21. Tugwood JD, Issemann I, Anderson RG, Bundell KR, McPheat WL, Green S. The mouse peroxisome proliferator activated receptor recognizes a response element in the 5' flanking sequence of the rat acyl CoA oxidase gene. EMBO J 1992;11:433-9.
22. Zhang B, Marcus SL, Sajjadi FG, Alvares K, Reddy JK, Subramani S, et al. Identification of a peroxisome proliferator-responsive element upstream of the gene encoding rat peroxisomal enoyl-CoA hydratase/3-hydroxyacyl- CoA dehydrogenase. Proc Natl Acad Sci U S A 1992;89:7541-5.
43
elements implicated in the regulation of CYP4A1 transcription. Biochem J 1995;306:473-9. 24. Vanden Heuvel JP. Peroxisome proliferator-activated receptors: a critical link among fatty acids, gene expression and carcinogenesis. J Nutr 1999;129:575S-80S.
25. Fajas L, Auboeuf D, Raspe E, Schoonjans K, Lefebvre AM, Saladin R, et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem 1997;272:18779-89.
26. Fajas L, Fruchart JC, Auwerx J. PPARgamma3 mRNA: a distinct PPARgamma mRNA subtype transcribed from an independent promoter. FEBS Lett 1998;438:55-60.
27. Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, et al. Ligand binding and co-activator assembly of the peroxisome proliferator- activated receptor-gamma. Nature 1998;395:137-43.
28. Juge-Aubry C, Pernin A, Favez T, Burger AG, Wahli W, Meier CA, et al. DNA binding properties of peroxisome proliferator-activated receptor subtypes on various natural peroxisome proliferator response elements. Importance of the 5'-flanking region. J Biol Chem 1997;272:25252-9.
29. DiRenzo J, Soderstrom M, Kurokawa R, Ogliastro MH, Ricote M, Ingrey S, et al. Peroxisome proliferator-activated receptors and retinoic acid receptors differentially control the interactions of retinoid X receptor heterodimers with ligands, coactivators, and corepressors. Mol Cell Biol 1997;17:2166-76.
30. Rosen ED, Spiegelman BM. PPARgamma : a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem 2001;276:37731-4.
44
31. Zhu Y, Alvares K, Huang Q, Rao MS, Reddy JK. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J Biol Chem 1993;268:26817-20.
32. Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996;137:354-66.
33. Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev 1999;20:649-88.
34. Wu Z, Xie Y, Morrison RF, Bucher NL, Farmer SR. PPARgamma induces the insulin-dependent glucose transporter GLUT4 in the absence of C/EBPalpha during the conversion of 3T3 fibroblasts into adipocytes. J Clin Invest 1998;101:22-32.
35. Brown KK, Henke BR, Blanchard SG, Cobb JE, Mook R, Kaldor I, et al. A novel N-aryl tyrosine activator of peroxisome proliferator-activated receptor-gamma reverses the diabetic phenotype of the Zucker diabetic fatty rat. Diabetes 1999;48:1415-24.
36. Reddy JK, Rao MS, Azarnoff DL, Sell S. Mitogenic and carcinogenic effects of a hypolipidemic peroxisome proliferator, [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio]acetic acid (Wy-14, 643), in rat and mouse liver. Cancer Res 1979;39:152-61.
37. Peters JM, Cattley RC, Gonzalez FJ. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis 1997;18:2029-33.
45 1996;87:159-70.
39. He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 1999;99:335-45.
40. Okano H, Shiraki K, Inoue H, Yamanaka T, Deguchi M, Sugimoto K, et al. Peroxisome proliferator-activated receptor gamma augments tumor necrosis factor family-induced apoptosis in hepatocellular carcinoma. Anticancer Drugs 2002;13:59-65.
41. DuBois RN, Gupta R, Brockman J, Reddy BS, Krakow SL, Lazar MA. The nuclear eicosanoid receptor, PPARgamma, is aberrantly expressed in colonic cancers. Carcinogenesis 1998;19:49-53.
42. Kilgore MW, Tate PL, Rai S, Sengoku E, Price TM. MCF-7 and T47D human breast cancer cells contain a functional peroxisomal response. Mol Cell Endocrinol 1997;129:229-35. 43. Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, et al. Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell 1998;1:465-70.
44. Kubota T, Koshizuka K, Williamson EA, Asou H, Said JW, Holden S, et al. Ligand for peroxisome proliferator-activated receptor gamma (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Res 1998;58:3344-52.
45. Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med 1998;4:1046-52.
46. Altiok S, Xu M, Spiegelman BM. PPARgamma induces cell cycle withdrawal: inhibition of E2F/DP DNA- binding activity via down-regulation of PP2A. Genes Dev
46 1997;11:1987-98.
47. Elstner E, Muller C, Koshizuka K, Williamson EA, Park D, Asou H, et al. Ligands for peroxisome proliferator-activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci U S A 1998;95:8806-11.
48. Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci U S A 1987;84:1005-9. 49. Kim HI, Cha JY, Kim SY, Kim JW, Roh KJ, Seong JK, et al. Peroxisomal proliferator-activated receptor-gamma upregulates glucokinase gene expression in beta-cells. Diabetes 2002;51:676-85.
50. Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A 1994;91:7355-9.
51. Spandau DF, Lee CH. Trans-activation of viral enhancers by the hepatitis B virus X protein. J Virol 1988;62:427-34.
52. Kim HI, Kim JW, Kim SH, Cha JY, Kim KS, Ahn YH. Identification and functional characterization of the peroxisomal proliferator response element in rat GLUT2 promoter. Diabetes 2000;49:1517-24.
53. Kim J, Chwae YJ, Kim MY, Choi IH, Park JH, Kim SJ. Molecular basis of HLA-C recognition by p58 natural killer cell inhibitory receptors. J Immunol 1997;159:3875-82. 54. Wu Q, Li Y, Liu R, Agadir A, Lee MO, Liu Y, et al. Modulation of retinoic acid
47
sensitivity in lung cancer cells through dynamic balance of orphan receptors nur77 and COUP-TF and their heterodimerization. EMBO J 1997;16:1656-69.
55. Jiang S, Song MJ, Shin EC, Lee MO, Kim SJ, Park JH. Apoptosis in human hepatoma cell lines by chemotherapeutic drugs via Fas-dependent and Fas-independent pathways. Hepatology 1999;29:101-10.
56. He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A 1998;95:2509-14.
57. Fajas L, Debril MB, Auwerx J. Peroxisome proliferator-activated receptor-gamma: from adipogenesis to carcinogenesis. J Mol Endocrinol 2001;27:1-9.
58. Altucci L, Gronemeyer H. Nuclear receptors in cell life and death. Trends Endocrinol Metab 2001;12:460-8.
59. Makino Y, Yoshikawa N, Okamoto K, Hirota K, Yodoi J, Makino I, et al. Direct association with thioredoxin allows redox regulation of glucocorticoid receptor function. J Biol Chem 1999;274:3182-8.
60. Moilanen AM, Poukka H, Karvonen U, Hakli M, Janne OA, Palvimo JJ. Identification of a novel RING finger protein as a coregulator in steroid receptor-mediated gene transcription. Mol Cell Biol 1998;18:5128-39.
61. Tsutsumi T, Suzuki T, Shimoike T, Suzuki R, Moriya K, Shintani Y, et al. Interaction of hepatitis C virus core protein with retinoid X receptor alpha modulates its transcriptional activity. Hepatology 2002;35:937-46.
48
receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell 1997;89:373-80.
63. Glass CK, Rose DW, Rosenfeld MG. Nuclear receptor coactivators. Curr Opin Cell Biol 1997;9:222-32.
64. Folkers GE, van der Saag PT. Adenovirus E1A functions as a cofactor for retinoic acid receptor beta (RAR beta) through direct interaction with RAR beta. Mol Cell Biol 1995;15:5868-78.
65. Sista ND, Barry C, Sampson K, Pagano J. Physical and functional interaction of the Epstein-Barr virus BZLF1 transactivator with the retinoic acid receptors RAR alpha and RXR alpha. Nucleic Acids Res 1995;23:1729-36.
66. Henkler F, Hoare J, Waseem N, Goldin RD, McGarvey MJ, Koshy R, et al. Intracellular localization of the hepatitis B virus HBx protein. J Gen Virol 2001;82:871-82. 67. Wang XW, Forrester K, Yeh H, Feitelson MA, Gu JR, Harris CC. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci U S A 1994;91:2230-4.
68. Mueller E, Smith M, Sarraf P, Kroll T, Aiyer A, Kaufman DS, et al. Effects of ligand activation of peroxisome proliferator-activated receptor gamma in human prostate cancer. Proc Natl Acad Sci U S A 2000;97:10990-5.
69. Tsubouchi Y, Sano H, Kawahito Y, Mukai S, Yamada R, Kohno M, et al. Inhibition of human lung cancer cell growth by the peroxisome proliferator-activated receptor-gamma agonists through induction of apoptosis. Biochem Biophys Res Commun 2000;270:400-5.
49
70. Butler R, Mitchell SH, Tindall DJ, Young CY. Nonapoptotic cell death associated with S-phase arrest of prostate cancer cells via the peroxisome proliferator-activated receptor gamma ligand, 15-deoxy-delta12,14-prostaglandin J2. Cell Growth Differ 2000;11:49-61.
50 국문요약
HBx 에 의한 PPAR
γ
의 전사활성 조절
최 윤 희 연세대학교 대학원 의과학사업단 <지도 김 세 종 교수> B 형 간염바이러스 X 단백질(HBx)은 여러 세포 내 단백질과 상호 결합함 으로써 그 생물학적 기능을 수행하며, 간암발생과정에 중요한 역할을 하는 것 으로 알려져 왔다. PPARγ는 핵 수용체의 한 종류로, 세포내의 대사과정, 세포의 증식과 사멸 및 종양화에 관여하는 전사인자이다. 본 연구에서는 HBx 가 PPARγ와 세포질 내에서 결합하여 핵으로의 translocation 을 차단함으로써, PPARγ의 전사활성과 세포고사 유도 기능을 억제함을 밝혔다. HBx 와 PPARγ가 상호결합함을 관찰하였고, 사람 간세포주인 Chang 세포에 HBx 와 PPARγ를 동 시에 발현시킨 결과, PPARγ가 HBx 와 co-localization 하며 세포질 내에 더 많이 존재하는 것을 확인함으로써, 정상적으로 주로 핵 내에 존재하는 PPARγ가 핵 내로 translocation 되지 못하는 것을 관찰할 수 있었다. 대조 마우스와 HBx 형 질전환 마우스에서 HBx 과발현에 따른 PPARγ의 발현량에는 차이가 없음에도 불구하고, 핵 내 존재하는 PPARγ는 HBx 형질전환 쥐에서 상당히 감소되어 있51
었다. 또한 HBx 는 PPARγ의 DNA 결합을 억제하였으며, 재조합 HBx 단백질을 핵단백질과 반응시켰을 때, HBx 농도 의존적으로 PPARγ의 DNA 결합능이 감소 되는 것을 확인하였다. PPARγ의 response element 를 포함하는 reporter 의 활성도 HBx 의 농도에 따라 현저히 억제됨을 관찰함으로써 HBx 에 의한 PPARγ 전사활
성억제를확인하였다. 세포증식을 억제하는 기능을 갖고 있는 것으로 알려진
PPARγ의 리간드인 troglitazone 을 사용하여 HepG2 와 HepG2.2.15 세포의 세포증 식을 비교해 본 결과, HepG2 세포에서는 troglitazone 에 의해 세포증식이 억제되 었음에 반해, HBV 가 형질 도입된 HepG2.2.15 세포에서는 이러한 증식억제 효 과를 볼 수 없었다. HepG2 세포주에 아데노바이러스를 이용한 HBx 이입시킨 결 과 또한 troglitazone 에 의한 세포증식억제가 유도되지 않았다. 이상의 결과로 HBx 는 PPARγ과 세포질 내에서 복합체를 이루어, PPARγ의 고유한 기능인 전사 활성 기능과 세포고사 유도 기능을 억제함을 확인하였고, 이러한 결과는 B 형 간염바이러스 감염으로 야기되는 간질환의 병인과정에 중요한 역할을 담당할 것으로 생각된다. 핵심되는 말 : HBx, PPARγ, 전사활성, 단백질−단백질간상호작용