이학 석사학위 논문
Hepatocyte Growth Factor Inhibits Glial Scar Formation
and Decreases Production of Chondroitin Sulfate
Proteoglycans after Spinal Cord Injury
아 주 대 학 교 대 학 원
의 생 명 과 학 과
Hepatocyte Growth Factor Inhibits Glial Scar Formation
and Decreases Production of Chondroitin Sulfate
Proteoglycans after Spinal Cord Injury
by
Soo Ryung Jeong
Major in Neuroscience
Department of Biomedical Sciences
Hepatocyte Growth Factor Inhibits Glial Scar Formation
and Decreases Production of Chondroitin Sulfate
Proteoglycans after Spinal Cord Injury
by
Soo Ryung Jeong
A Dissertation Submitted to The Graduate School of Ajou University
in Partial Fulfillment of the Requirements for the Degree of
Master of Neuroscience
Supervised by
Byung Gon Kim, M.D., Ph.D.
Major in Neuroscience
Department of Biomedical Sciences
Graduate School, Ajou University
The Graduate School, Ajou University
ACKNOWLEDGEMENT
참으로 길게만 보였던 졸업이 어느덧 가까이 다가왔네요. 간절히 바랬던 이 순간이 다가오니 지나온 시간들이 눈앞을 스칩니다. 내가 쓴 논문 한편이 갖고 싶어서 시작했던 석사생활… 그 짧았던 이 년 동안, 삶에서 중요한 결혼을 했고, 외국학회도 가고, 포스터 발표도 하고.. 바쁘게 뛰어다녔던 시간이었습니다. 끝나지 않을 것만 같던 시간이 흘러 어느덧 논문이 나오게 되니 이 순간이 있기까지 저를 도와주셨던 많은 분들이 생각납니다. 우선 모든 부분들을 꼼꼼히 지도해 주신 김병곤 교수님께 진심으로 감사 드립니다. 시간이 흐를수록, 교수님이 저의 지도교수님이라는 사실이 너무나도 자랑스럽습니다. 교수님 감사합니다. 그리고 저의 부족한 부분을 채워주시고 양리학 교실에서 실험을 배울 수 있게 해주셨던 조은혜 교수님께도 감사말씀 전합니다. 또한, 공개발표 때 아낌없는 충고해주신 이명애 교수님, 감사합니다. 세포를 공급해주신 해부학 교실에 서해영 선생님과 김성수 선생님, 그리고 스케줄에 맞춰 준비해주신 다영언니 에게도 감사 드립니다. 그리고 무엇보다 즐거운 추억이 가득한 석사생활을 함께 한 실험실 식구들 감사합니다. 동훈오빠! 오빠가 있어서 제 실험실생활이 너무나도 윤택했어요. 그리고 혁민오빠! 무신경 한 듯 하지만 누구보다도 잘 챙겨주고, 잘 가르쳐주신 오빠. 오빠에게서 실험뿐만 아니라, 인생사는 법도 배웠어요. 영미! 기쁠 때 슬플 때 항상 내 곁에 있고, 도와주고 힘을 줬던 영미. 우리방 막내 민정이! 넌 실험실에 활력소였다. 거의 우리방 식구인 경진언니, 범수오빠, 석순오빠. 여러분들 덕분에 너무나 행복했습니다. 마지막으로 처음엔 반대했지만 끝까지 아낌없는 사랑으로 저를 지지해주고, 도와주신 부모님과 석사를 시작할 수 있도록 용기를 주고, 졸업할 수 있도록 내조해준 남편… 고맙습니다 그리고 사랑합니다.- ABSTRACT -
Hepatocyte Growth Factor Inhibits Glial Scar Formation and
Decreases Production of Chondroitin Sulfate Proteoglycans after
Spinal Cord Injury
Formation of glial scars physically impedes growth of regenerating axons following CNS injuries such as spinal cord trauma. Glial scars also produce various species of chondroitin sulfate proteoglycans (CSPGs) that act as a potent inhibitor of axon growth. Thus, inhibition of glial scar formation and/or suppression of CSPGs production may support axonal regeneration following spinal cord injury. Hepatocyte growth factor (HGF), which was originally identified as a mitogen for hepatocytes, has been shown to strongly suppress fibrosis of internal organs such as liver and kidney. However, its role in the regulation of CNS glial scars has not been studied. The purpose of this study was to examine whether HGF can affect formation of glial scars and production of various CSPGs. Glial scar formation was mimicked in vitro by cytokine treatment in primary astrocyte culture. Treatment of TGFβ1 led to astrocytic hypertrophy that was associated with dramatic upregulation of intermediate filament, GFAP (glial fibrillary acidic protein), which was evidently suppressed by HGF cotreatment in a dose-dependent manner. HGF also inhibited TGFβ1-induced upregulation of neurocan, one of CSPG species. To explore whether glial scar formation is inhibited by HGF in vivo spinal cord injury model, HGF overexpressing mesenchymal stem cells (HGF-MSCs) were transplanted into hemisected spinal cord lesions
at T8. The ex-vivo gene delivery approach effectively increased the HGF level and phosphorylation of its cognate receptor c-Met. Transplantation of HGF-MSCs reduced the formation glial scar and CSPGs deposition around the hemisection lesions. Western blot showed that the amount of neurocan significantly decreased in animals with HGF-MSCs transplantation. These findings were accompanied by decreases in TGFβ1 and β2, strong inducers of astrocyte activation. Our results suggest that HGF may potently regulate both glial scar formation and CSPGs production following spinal cord injury
Key words: spinal cord injury, hepatocyte growth factor, glial scar, chondroitin sulfate
TABLE OF CONTENTS
ABSTRACT ··· ⅰ TABLE OF CONTENTS ··· ⅲ LIST OF FIGURES ··· ⅴ LIST OF TABLE ··· ⅵ LIST OF ABBREVIATION ··· ⅶ Ⅰ. INTRODUCTION ··· 1Ⅱ. MATERIAL AND METHODS ··· 5
A. PRIMARY ASTROCYTE CULTURES ··· 5
B. ANIMALS AND SURGICAL PROCEDURES ··· 6
C. IMMUNOCYTOCHEMISTRY ··· 7
D. TISSUE PROCESSING AND IMMUNOHISTOCHEMISTRY ··· 7
E. ENZYME-LINKED IMMUNOSORBENT ASSAY (ELISA) ··· 8
F. WESTERN BLOT ANALYSIS ··· 9
G. REVERSE TRANSCRIPTION POLYMERASE CHAIN REACTION (RT-PCR) ··· 11
H. STASTITATIC ANALYSIS ··· 12
Ⅲ. RESULTS ··· 13
A. HGF prevents cytokine-induced astrocytic activation in vitro. ··· 13
B. HGF decreases expression of chondroitin sulfate proteoglycans (CSPGs) ··· 19
D. Ex vivo delivery of HGF using mesenchymal stem cells in hemisection spinal
cord injury. ··· 27
E. HGF inhibits astrocytic activation and CSPGs production after spinal cord injury. ··· 33
Ⅳ. DISCUSSION ··· 38
Ⅴ. CONCLUSION ··· 43
REFERENCE ··· 44
LIST OF FIGURES
Fig. 1. Astrocytic activation in vitro glial scar model ··· 15
Fig. 2. Absence of HGF effect on astrocyte proliferation ··· 17
Fig. 3. Expression of HGF receptor, c-Met, in primary astrocytes ··· 18
Fig. 4. Modulation of chondroitin sulfate proteoglycans (CSPGs) mRNA expression by HGF ··· 20
Fig. 5. HGF inhibited secretion of TGFβ1 and TGFβ2 from activated astrocytes. ··· 24
Fig. 6. Ex vivo delivery of HGF gene using human mesenchymal stem cells as carriers ·· 29
Fig. 7. Verification of effective HGF delivery by the ex vivo method ··· 30
Fig. 8. Reduction of astrocytic scars by transplantation of HGF overexpressing mesenchymal stem cells ··· 35
Fig. 9. Regulation of TGFβ1 and TGFβ2 level in glial scars by transplantation of HGF-MSCs ··· 36 Fig. 10. Reduction of CSPGs production by HGF-MSCs in spinal cord injury model ···· 37
LIST OF TABLES
Table 1. Experimental groups and treatment number in vitro ··· 21
Table 2. Experimental groups and treatment number of IL-1β and IFNγ-induced in
vitro scar model ··· 26
Table 3. HGF concentration in cultured supernatant by culture period condition · 31
LIST OF ABBREVIATION
c-Met, mesenchymal-epithelial transition factor / hepatocyte growth factor receptor CNS, central nervous system
CS-56, anti-chondroitin sulfate
CSPG, chondroitin sulfate proteoglycan DAPI, 4',6-Diamidino-2-Phenylindole DW, distilled water
ECM, extracellular matrix
ELISA, enzyme-linked immunosorbent assay EtBr, ethidium bromide
FBS, fetal bovine serum
GFAP, anti-glial fibrillary acidic protein HGF, hepatocyte growth factor
Hu-Mito anti-human mitochondria IFNr, interferon ramma
IL1b, interleukin 1 beta
MEM/EBSS, minimum essential medium with Earle's Balanced Salts MOI, multiplicity of infection
MSC, mesenchymal stem cell NGS, normal goat serum P/S, penicillin streptomycin PNN, perineuronal net
PBS, phosphate buffered saline PFA, paraformaldehyde PVDF, polyvinylidene difluoride RT, room temperature SCI, spinal cord injury TBS, tris-bufferd saline
TGFβ1, transforming growth factor beta 1 TGFβ2, transforming growth factor beta 2
Ⅰ. INTRODUCTION
Traumatic spinal cord injury is usually accompanied by development of cystic cavities surrounded by glial scars which severely impair regeneration of severed axons (Sandvig et al., 2004; Silver and Miller, 2004; Yiu and He, 2006). Glial scars are composed of various types of cells and extracellular matrices. Of these, astrocytes play key roles in the formation of glial scars. Upon injury, they upregulate the expression of intermediate filament proteins (known as glial fibrillary acidic proteins, GFAP) to become hypertrophied (Pekny and Nilsson, 2005). These reactive astrocytes also secret various extracellular matrix (ECM) proteins and form dense physical barriers together with ECM molecules. Although glial scars may exert some beneficial effects by regulating local immune responses and promoting tissue repair (Faulkner et al., 2004; Rolls et al., 2009), mounting evidence indicates that glial scars constitute a major component of post-injury environment unfavorable to spontaneous axonal regeneration (Yiu and He, 2006).
Formation of astrocytic glial scars impedes growth of regenerating axons both physically and chemically. Hypertrophic astrocytes pose mechanical barriers to growing axons (McKeon et al., 1991). Deletion of both GFAP and vimentin, major intermediate filaments of astrocytes upregulated after CNS injury, resulted in enhancement of axonal regeneration after spinal cord injury (Ribotta et al., 2004). At the same time, they also produce growth inhibitory extracellular matrix (ECM) molecules such as chondroitin sulfate proteoglycans (CSPGs) (McKeon et al., 1999; Tang et al., 2003). CSPGs, potent inhibitory ECM molecules, consist of a core protein and many sugar side chains, which are large, sulfated
glycosaminoglycans (GAG) chains covalently attached to core proteins (Smith and Strunz, 2005; Galtrey and Fawcett, 2007). There are several different CSPG species upregulated after SCI such as neurocan, brevican, phosphacan, and NG2 proteoglycan (Jones et al., 2003; Tang et al., 2003). Regardless of different classes of core proteins, major inhibitory functions appear to reside in GAG chains. Several studies have reported that enzymatic degradation of GAG chains by chondroitinase ABC enhances axonal regeneration and improves functional recovery after SCI (Bradbury et al., 2002; Caggiano et al., 2005; Tester and Howland, 2008; Tom and Houle, 2008). These studies highlight importance of manipulating reactive astrocytes and ECM proteins to enhance regeneration and functional recovery after SCI.
Previous studies have shown that various inflammatory cytokines are implicated in induction of reactive astrocytosis. Especially, early induction of TGFβs after SCI is considered to be essential in reactive astrocytosis (Logan et al., 1994; Reilly et al., 1998; Fitch et al., 1999; Gomes et al., 2005; Buss et al., 2008). Treatment of neutralizing antibodies against TGFβs led to marked attenuation of CNS scarring, suggesting their causative role (Logan et al., 1999; Moon and Fawcett, 2001). In addition to TGFβs, however, many other factors are implicated in activation of astrocytes at least in vitro. Several proinflammatory cytokines such as IL-1β, IFNγ and TNFa (Hewett et al., 1993; John et al., 2003) also strongly induce astrocytosis. Furthermore, it has been shown that nitric oxide (NO) (Brahmachari et al., 2006), oxidative stress (Rohl et al., 2008) and ciliary neurotrophic factor (CNTF) (Hudgins and Levison, 1998; Levison et al., 1998) could elicit reactive astrocytosis. Therefore, it would be very challenging to identify a specific target of which blockade could effectively prevent reactive astrocytosis.
Hepatocyte growth factor (HGF) was first discovered and cloned as a mitogenic polypeptide for hepatocytes (Nakamura et al., 1989). HGF exerts its actions through tyrosine kinase receptor, c-Met (Shimamura et al., 2007). Beyond its classical effects on hepatocytes, subsequent studies have uncovered diverse functions of HGF in nervous system. It has a crucial role in developing neurons; HGF promotes neurite outgrowth from motor neuron (Ebens et al., 1996; Yamamoto et al., 1997) and improves neuron survival (Maina and Klein, 1999). It also stimulates proliferation and migration of Schwann cells (Krasnoselsky et al., 1994), olfactory interneuron precursors (Garzotto et al., 2008), and oligodendrocyte precursor cells (Yan and Rivkees, 2002; Ohya et al., 2007). One of the well established functions of HGF outside CNS is to suppress fibrosis of internal organs such as the liver and kidney (Matsumoto and Nakamura, 1992). It has been shown that liver fibrosis is actively processed by TGFβ1 (Border and Noble, 1994) and inhibition of TGFβ1 prevents the progression of liver fibrosis and even improves hepatocyte regeneration (Nakamura et al., 2000). HGF also functions to suppress fibrosis of kidney by inhibiting TGFβs production (Liu, 2004, 2006; Liu and Yang, 2006). Regulation of proteoglycans synthesis (Kobayashi et al., 2003) and extracellular matrix remodeling by HGF further contributed to attenuation of fibrosis in the internal organs (Kim et al., 1997; Liu and Yang, 2006). Interestingly, formation of fibrosis in the liver or kidney shares similarity with glial scarsring in CNS in that TGFβs are critically involved and deposition of ECM constitute an important component. Based on this reasoning, it could be hypothesized that HGF may play a role in modulation of CNS glial scars. However, the possibility that HGFs may play an inhibitory role in the glial scars formation has not been addressed to date.
The purpose of this study was to examine whether HGFs can affect formation of glial scars and the production of various CSPGs. We found that HGF prevented both a formation of astrocyte activation and a production of chondroitin sulfate proteoglycans in activated astrocyte culture system, suggesting that HGFs may regulate scar formation. These effects appeared to be mediated by decreasing the level of TGFβ1. HGF’s effects on glial scars were also demonstrated in vivo by transplantation of HGF overexpressing mesenchymal stem cells into hemisected spinal cord tissue. Our data suggest that HGFs evidently regulate both scar formation and production of CSPGs following spinal cord injury.
Ⅱ. MATERIALS AND METHODS
A. Primary astrocyte cultures
Primary astrocyte cultures from cerebral cortex were prepared from postnatal 1 day pups as described previously (Wilson and Dixon, 1989). The upper part of the skull was separated and the meningeal tissue removed. The cerebral neocortices were isolated, placed into complete medium, which were containing 10% fetal bovine serum (FΒS), 0.01 M HEPES, 1% penicillin streptomycin in MEM/EΒSS (Minimum Essential Medium with Earle's Balanced Salts, Hyclone). The neocortices were homogenized by pipetting, were plated onto 175 cm2 tissue culture flasks (Corning). The cultures were maintained in a humidified atmosphere of 95% air 5% CO2 at 37℃ for 10~14 days after plating. Flasks were
slapped to detach nonadherent cells from the bottom of flasks and then were washed with PΒS. Adherent astrocytes were removed by treatment with trypsin (0.25%, Gibco), were resuspended in complete medium, and suspended cells properly seeded in each condition. Then microglia and oligodendrocytes were extracted. Once cells reached confluence (5-7 days), they were plated into 100 mm2 (Falcon) at a density of 1.3 X 106 cells/dish. Cells were passaged again and plated. The final percentage of GFAP-expressing cells in these cultures was found to be >95%. When the cells reached confluence, culture medium was changed to serum-free MEM containing 1 % penicillin streptomycin (P/S) and 0.01M HEPES before cytokine treatment. Astrocytes were treated in serum free medium with IL-1β (R&D systems) 10 ng/ml and IFNγ (R&D systems) 10 ng/ml, or TGFβ1 (Peprotech) 10 ng/ml with or
without HGF (Millipore) of various concentrations for 24 hours.
B. Animals and surgical procedures
Adult female Sprague Dawley rats weighing 200-250 g were used in this study. SD rats were housed in the Ajou University Animal Care and Use Committee. For surgery, animals were anaesthetized with chloral hydrate (400mg/kg, i.p.), received a dorsal laminectomy to expose the spinal cord. The dura were opened and iridectomy scissors were used to create a spinal cord right-over hemisected injury at Thoracic 8 level. Vacuum suction was used to aspirate the remaining tissues. Transplants were prepared with the concentration of 4.0 X 104 cells/ul and 5 μl of cell suspensions (total 2.0 X 105
cells) were soaked into gelfoam pledgets. The gelfoams were immediately implanted into right-hemisection lesions. And then rats by each transplantation groups were injected 4 sites in rostral and caudal of injured spinal cord using a hamilton syringe. Each site was given 1 ul of transplant which is prepared 4 X 104 cells/ul concentrations. So, rats totally transplanted 3.6 X 105 cells / 9ul by concentration. Experimental groups were divided into control group with phosphate buffered saline (PΒS) soaked gelfoam, MSC only group, and HGF-MSC group with gelfoams soaked with HGF overexpressing MSCs. All animals received daily intraperitoneal cyclosporine (NORVATIS) at a dosage of 10 mg/kg beginning from one day before transplantation to sacrifice.
Prophylactic antibiotics (
cefazolin)were intraperitoneally injected on the next day
after each surgery, and a bladder care had been provided twice daily until the animals
resumed self-voiding.
C. Immunocytochemistry
For immunocytochemistry, cells were plated on 9 mm or 12 mm coverslips coated with poly-D-lysine (Sigma). Attached cells on coverslip were fixed in 4% paraformaldehyde (PFA) for 30min at room temperature (RT) after 3 times PΒS washing each for 10 minutes. After blocking with 10% normal goat serum for 1hr, primary antibodies were applied in the same blocking solution at 4℃ overnight or for 4 hours at RT. After thorough washing with PΒS, appropriate secondary antibodies tagged with Alexa Fluor 488 or Alexa Fluor 594 (Molecular Probes, Eugene, OR) were applied for 1hr at RT. The primary antibodies used in this study were mouse anti-GFAP (Dako; 1:400), rabbit anti-c-met (Santa Cruz; 1:100), rabbit anti-phospho-c-Met (Invitrogen; 1:100), mouse anti-CS-56 (Sigma; 1:400), mouse anti-neurocan (Millipore; 1:100), mouse anti-human mitochondria (Chemicon; 1:400) and rabbit anti-Ki67 (novocasta; 1:400). The coverslips were mounted onto slides with glycerol based mounting medium (Biomeda, Foster City, CA). The images were taken using a FV 300 confocal microscope (Olympus, Tokyo, Japan). These experiments are repeated several trials by experiment number of each group as described the table 1.
D. Tissue processing and immunohistochemistry
Rats were anesthetized with an overdose of chloral hydrate and perfused with heparinized saline (0.9%) followed by 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer. The spinal cord containing lesion site was dissected and post-fixed in 4% PFA for 2 hours, followedby cryoprotection in a graded series of sucrose solutions. Cryosections for
spinal cord (20 μm thickness) were made longitudinally in a 1:10 series, mounted onto Super Frost plus slides (Fisher Scientific, Pittsburgh, PA), and stored at -20℃ until use. For immunohistochemistry, longitudinal tissue sections were treated with 10% normal goat serum (Hyclone, Logan, VT) to prevent nonspecific immunoreactivity. Primary antibodies dissolved in the same blocking solution were applied onto tissue sections at 4℃ overnight followed by appropriate secondary antibodies tagged with Alexa Fluor 488 or Alexa Fluor 594 (Molecular Probes, Eugene, OR) for 1hr at RT. The primary antibodies used for immunohistochemistry were same as those for immunocytochemistry. To quantification of fluorescence intensity, such as GFAP and CS-56, we analyzed confocal images of 3 sites that are rostral, caudal and contra lateral beyond the glial limitance from 3 cryo-section at each group.The images were taken using a FV 300 confocal microscope (Olympus, Tokyo, Japan). Immunohistochemistry is repeated several trials by experiment number of each group as described the table 4.
E. Enzyme-Linked Immunosorbent Assay (ELISA)
Cultured supernatants or spinal cord lysates were subjected to ELISA analysis to determine the level of TGFβs and HGF. For cultured supernatants, primary astrocyte cultures were treated with cytokines with or without HGF (as described above) for 24 hours and supernatants were collected. Tissue samples were homogenized with a dounce tissue grinder in ice-cold homogenization buffer (50mM Tris-HCl, pH 7.6, containing 150 mM NaCl, 1mM EDTA, 0.32M sucrose) supplemented with 1 homogenized with a douncetor cocktail (Roche,
Mannheim, Germany). The homogenates were then centrifuged at 12,000 rpm for 10 min and the supernatants were used for ELISA analysis. Protein concentration of the cultured supernatants or tissue homogenates were measured using Micro BCA Protein Assay kit (Pierce, Rockford, IL), and equal amounts of samples (typically 10 to 20 µg) were loaded into 96 well plates coated with capture antibodies. Concentration of HGF was assayed using human HGF ELISA kit (immunis, EIA, Japan) and that of TGFβ1 and β2 using kits from R&D systems (MN, USA). Detailed procedures followed an instruction manual provided by corresponding manufacturers. ELISA is tried by experiment number of each group as described the table 2 and table 4.
F. Western Blot analysis
One week after surgery, 17 rats were anesthetized with overdose chloral hydrate and briefly perfused with ice-cold saline to remove blood cells. Five mm-long spinal cord blocks containing lesion site were quickly dissected and homogenized in tissue extraction buffer containing protease inhibitor cocktail (Pierce) and phosphatase inhibitor (Halt Phosphatase Inhibitor Cocktail, Themo), which safeguards against serine, threonine and protein tyrosine phosphatase activities, on ice. Protein concentration was determined using the BCA protein Assay kit (Pierce). Equal amounts (24 ug / 30ul) of proteins were analyzed by SDS-PAGE, together with size marker (Thermo, Rockford, IL). It is performed in 10% polyacrylamide gels with 8% stacking gels. Proteins were separated by constant voltage of 130 mV in RT after separated by 60 mV in stacking gels and transferred to polyvinylidene difluoride
(PVDF) membranes (Immobilon-P; Millipore, Bedford, MA). The membranes were washed in Tris-buffered saline (TΒS-, 0.9% NaCl, 10mM Tris-HCl pH 8.0) containing 0.1% Tween-20 (Sigma) (TΒS-T) and blocked 5% skim milk for 2 hours at RT. The membranes were then incubated with following primary antibodies in 5% ΒSA solution. : anti-GFAP (Dako) and anti-c-met (SantaCruz, 1:1000). Especially anti-phospho-c-met (Invitrogen, 1:1000) were used in 1% ΒSA solution. β-actin was used as a loading control for cell lysates After incubation with horseradish peroxidase conjugated secondary antibodies, immunoreactivity was visualized by chemiluminescence reagents (ECL advance western blot detection kit, GE healthcare).
For neurocan detection, tissue lysates were treated with 0.03 U chondroitinase ABC (protease-free, SEIKAGAKU), which is cut the GAG chain from core protein, for 2 hours at 37℃. SDS-PAGE was performed in 6% polyacrylamide gels with a 4% stacking gel. Proteins were prepared by 24 ug/30ul concentration and separated by constant voltage of 60 mV in RT after separated by 40 mV in stacking gels. Separated proteins in gel were transferred to PVDF membranes (Millipore) at 4℃ for 15 hours by constant current of 150mA in cold room. The blot membrane was rinsed three times in Tris-buffered saline (TΒS, 0.9% NaCl, 10mM Tris-HCl pH7.5) containing 0.05% Tween 20 (Sigma) (TΒS-T) and blocked 5% skim milk for 2 hours at RT. The blot was incubated with anti-neurocan (Millipore) in 5% skim milk solution. Anti-neurocan antibody can detect the 1D2 (150-163 kDa, C-terminal epitope of neurocan, ), 1F6 (122-130 kDa, N-terminal epitope of neurocan) and intact neurocan (240-270kDa) (Asher et al., 2000). After incubation with horseradish peroxidase conjugated secondary antibodies dissolving in 5% skim milk, immunoreactivity
was visualized by chemiluminescence reagents (ECL advance western blot detection kit, GE healthcare). This analysis is repeated several trials by experiment number of each group as described the table 4
G. Reverse Transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted from cultured cells using Trizol (Gibco) according to the manufacturer’s protocol. The amount of RNA was determined using spectoscopy at 260 nm. Five ug of RNA was reverse transcribed to cDNA using a standard RT protocol. One ul (0.4 ug) of cDNA was added to PCR-reaction premix (GenDEPOT) with 10 pM corresponding primer pairs. The following primers were used for polymerase chain reaction : GAPDH, 5’-GTG TAG TTC ACG CCC ACG TC-3’ (forward), 5’-5’-GTG ATG GCA TGG ACT 5’-GTG GT-3’ (reverse) ; neurocan, 5’-GCC ACA CTC TAC ACT CGT CCC-3’ (f), 5’TCT CCC CAG CAT AGC CCT GAT-3’(r) ; phosphacan, 5’-GCA AGT CCT GCC GTC CTT GCA 3’-(f), 5’-GGA ATA GGG ATT AGT AAC AGC-3’ (r) ; c-Met, 5’-TGT CTC TGA AAT CCA CCC GA 3’ (f), 5’ GTG TAG TTC ACG CCC ACG TC-3’ (r). Their specificity was verified using the basic local alignment search tool (BLAST) on the GenBank database. PCR amplification was performed with 35 cycles of 95 ℃ for 30 s, 55 ℃ ~ 60 ℃ (property Tm) for 30 s, 72 ℃ for 90 s. The PCR products were separated on a 1% agarose gel and stained by Ethidium Bromide (EtBr). The amount of each product was quantified by a gel document system (Bio-Rad). GAPDH expression was used as an internal reference to verify equal concentrations of cDNA in each sample. We have executed experimental number of each group as described
the table 1. And we quantified mRNA expression level of neurocan and phosphacan by band thickness using the image J program based on band thickness of non-TGFβ1 treatment.
H. Statistical Analysis
Statistical comparison of mean values was performed using one-way ANOVA followed by Tukey’s post hoc tests. All values are expressed as mean ± SD. Quantitation graphs were generated by GraphPad Prism version 4.00 (GraphPad Software, San Diego, CA, USA)
Ⅲ. RESULTS
A. HGF prevents cytokine-induced astrocytic activation in vitro.
To examine whether HGF affects formation of glial scars, we used primary cultured astrocytes isolated from 1 day pub rat’s neocortex to mimic glial scars (Fig. 1A). When more than 95% cultured astrocytes express glial fibrillary acidic protein (GFAP), we induced astrocyte activation by treatment of TGFβ1 at a concentration of 10 ng/ml (Baghdassarian et al., 1993; Reilly et al., 1998) (Fig. 1B, C). Immunocytochemistry revealed that TGFβ1-treated astrocytes upregulated GFAP positive intermediate filaments and showed hypertrophic morphology. HGF was added to the medium at three different concentrations: 5 ng/ml, 50 ng/ml, and 250 ng/ml. Fifty ng/ml and 250 ng/ml treatment of HGF obviously prevented morphological change, although 5 ng/ml was not effective (Fig. 1E-G). To quantify changes in astrocytic morphologies, we measured mean GFAP positive areas (Fig. 1G). We found that treatment of TGFβ1 clearly increased the mean size of astrocytes and co-treatment of HGF at 50 ng/ml and 250 ng/ml significantly reduced the extent of hypertrophic changes.
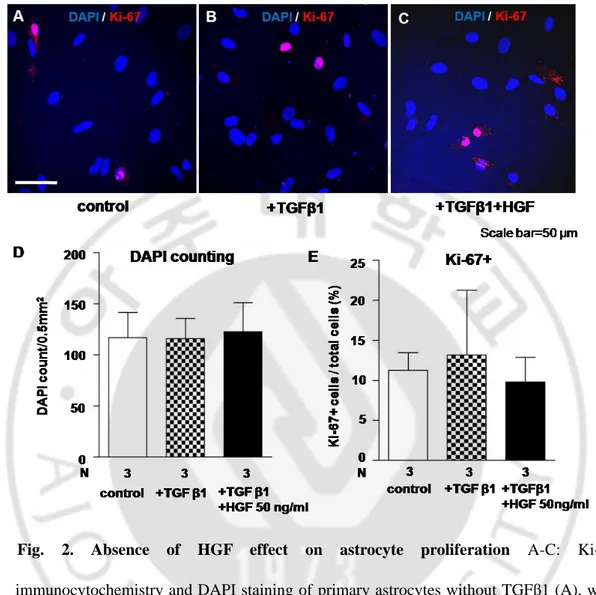
It has been previously reported that HGF stimulated neuronal cell survival and oligodendrocyte precursor cell proliferation (Yan and Rivkees, 2002; Ohya et al., 2007). To examine whether HGF affects proliferation of astrocytes, cells were stained with anti Ki-67, which is a cellular marker of proliferation and detects nuclei-during interphase. Ki-67 immunoreactivity was not different between different treatment groups (Fig. 2A-C).
Quantification of the number of Ki-67 positive cells showed that proliferation between TGFβ1 treatment and HGF treatment with TGFβ1 was not significantly different (Fig. 2E). As shown in Fig 2D, DAPI counts in regular area made no difference between TGFβ1 and HGF treatment, including control.
We next examined expression of HGF receptor c-Met in primary astrocyte. RT-PCR showed that c-Met mRNA is present in primary astrocytes and its expression is greatly increased after TGFβ1 treatment (Fig. 3A). HGF treatment did not change expression of c-Met. By immunostaining with antibodies that detect the beta-chain of c-Met (Fig. 3B-D), we confirmed the expression of c-Met in HGF treated astrocytes.
Fig. 1. Astrocytic activation in vitro glial scars model A: Illustration of primary astrocyte cultures and experimental scheme. B: GFAP stained astrocytes in serum free medium. B: 10 ng/ml treatment of TGFβ1 induced astrocytic hypertrophy. C: 5ng/ml treatment of HGF. D-F:5 ng/ml, 50 ng/ml, and 250 ng/ml HGF was added to culture medium with TGFβ1 respectively. Cells were treated with the above conditions for 24 hours and were fixed for immunocytochemistry with GFAP. G: Quantification of GFAP positive areas. *p<0.05, **p<0.01 and ***p<0.001 by one-way ANOVA followed by Tukey’s post hoc test for the comparison of mean GFAP areas between different groups.
Fig. 2. Absence of HGF effect on astrocyte proliferation A-C: Ki-67 immunocytochemistry and DAPI staining of primary astrocytes without TGFβ1 (A), with TGFβ1 (B), and TGFβ1 with HGF 50 ng/ml (C). D: DAPI counting results by groups. E: Quantification of Ki-67 positive cells counts among the all strocytes in the one taken picture.
Fig. 3. Expression of HGF receptor, c-Met, in primary astrocytes A: RT-PCR analysis showed mRNA expression of c-Met in primary astrocytes with or without TGFβ1 and HGF contreatment condition. B-D: Images of immunocytochemistry with c-Met and DAPI in primary astrocytes treated with TGFβ1 and HGF 50 ng/ml. B: Immunoreactivity of DAPI. C. Immunoreactivity of c-Met. D: merge image of DAPI and c-Met immunoreactivities.
B. HGF decreases expression of chondroitin sulfate proteoglycans (CSPGs)
To examine whether HGF affects production of CSPGs that is a major component of glial scars, we measured mRNA expression of neurocan and phosphacan, which are two species of CSPGs upregulated following cytokine stimulation (McKeon et al., 1999; Asher et al., 2000) and spinal cord injury (Jones and Tuszynski, 2002). TGFβ1 induced astrocytic activation resulted in dramatic increases in neurocan and phosphacan mRNAs. Quantification data showed that neurocan mRNA expression was elevated 8 times higher than control level. HGF treatment at a concentration of 50 ng/ml almost completely blocked the increase of neurocan mRNA expression (Fig. 4A, B). Phosphacan mRNA level was also increased 2 times with TGFβ1, and HGF co-treatment evidently reduce the expression level (Fig. 4C, D). These experiments confirmed that HGF obviously suppresses mRNA production of CSPGs.
Fig. 4. Modulation of chondroitin sulfate proteoglycans (CSPGs) mRNA expression by HGF A: Quantification of mRNA expression of neurocan. B: neurocan RNA analysis by RT-PCR. C: Quantification of mRNA expression of phosphacan D: phosphacan RNA analysis by RT-PCR. *p<0.05, **p<0.01 , and ***p<0.001 by one-way ANOVA followed by Tukey’s post hoc test for the comparison with hypertrophic effects in primary astrocytes.
Table 1. Experimental groups and treatment number in vitro Immunocytochemistry (GFAP) groups number Non- TGFβ1 (control) 4 TGFβ1 10 ng/ml 4 TGFβ1 10 ng/ml + HGF 5 ng/ml 3 TGFβ1 10ng/ml + HGF 50 ng/ml 4 TGFβ1 10 ng/ml + HGF 250 ng/ml 3
Immunocytochemistry, RT-PCR (c-Met) & Immunocytochemistry (Ki-67/DAPI)
groups number Non- TGFβ1 (control) 3 TGFβ1 10 ng/ml 3 TGFβ1 10ng/ml + HGF 50 ng/ml 3 RT-PCR (neurocan, phosphacan) groups number Non- TGFβ1 (control) 4 TGFβ1 10 ng/ml 4 TGFβ1 10ng/ml + HGF 50 ng/ml 4
C. Effects of HGF on TGFβ secretion
Following spinal cord injury, TGFβ1 and TGFβ2 were secreted from reactive astrocytes around the lesion site. TGFβ1 induces inflammatory responses and is involved in initial formation of the glial scars, while TGFβ2 maintains glial scars at later time points (Buss et al., 2008). Inhibition of TGFβ1 and TGFβ2 functions by neutralizing antibodies reduced the extent of scar formation (Logan et al., 1999; Moon and Fawcett, 2001). Therefore, TGFβs are regarded as strong inducers for glial scars formation.
For these reasons, we determined whether reduction of astrocytic activation is associated with changes in secretion of TGFβs. To this end, we adopted a different activation scheme. Treatment of interleukin-1 beta 1 (IL-1β) and interferon-gamma (IFNγ) has been shown to strongly induce astrocytic activation (Hewett et al., 1993). Applying IL-1β and IFNγ together is known to increase GFAP expression by NO induction (Brahmachari et al., 2006).
Purified astrocytes were treated with 10 ng/ml of IL-1β and 10 ng/ml of IFNγ for 48 hours after starvation during 24 hours (Fig. 5A). Treatment of IL-1β and IFNγ together elicited dramatic changes in astrocytic morphology (Fig 5B-D). Astrocytes that were exposed to IL-1β and IFNγ showed highly elongated morphology and tended to aggregate together compared to untreated astrocytes (Fig 5C). HGF treatment partially restored astrocytic morphology (Fig 5E). To compare secretion level of TGFβ1 and TGFβ2 from astrocytes cultured supernatants were harvested and analyzed for TGFβ1 and TGFβ2 ELISA after 48 hours of cytokines addition (Fig 5A, F-G). TGFβ1 secretion was significantly increased by IL-1β and IFNγ, which was completely blocked by HGF co treatment (Fig 5D).
TGFβ2 secretion is not significantly increased by addition of IL-1β and IFNγ, but clearly decreased by HGFs (Fig 5E). Thus, HGF inhibited secretion of TGFβ1 and TGFβ2 in IL-1β / IFNγ induced activated astrocyte culture system.
Fig. 5. HGF inhibited secretion of TGFβ1 and TGFβ2 from activated astrocytes. A: Schematic illustration of the experimental procedure. B-D: Representative images of GFAP Immunocytochemistry of primary astrocyte without cytokine treatment (B), with IL-1β and IFNγ at 10 ng/ml each for 48 hours (C), and addition of 50 ng/ml of HGF with IL-1β and IFNγ treatment (D). E-F: ELISA analysis of TGFβ1 (E) and TGFβ2 (F) in supernatants collected from cultures for 48 hours under designated experimental conditions. *p<0.05, and ***p<0.001 by one-way ANOVA followed by Tukey’s post hoc test for the comparison of TGFβ1 and TGFβ2 concentration between different groups.
Table 2. Experimental groups and treatment number of IL-1β and IFNγ-induced in vitro scar model
ELISA (TGFβ1 and TGFβ2)
groups number
Non IL-1β and IFNγ (control) 4 IL-1β 10 ng/ml + IFNγ 10 ng/ml 4 IL-1β 10 ng/ml + IFNγ 10 ng/ml + HGF 5 ng/ml 4
D. Ex vivo delivery of HGF using mesenchymal stem cells in hemisection spinal cord injury
In vitro data demonstrated that HGF prevents cytokine-induced astrocytic hypertrophy
and reduced CSPGs expression. We attempted to test whether HGF could regulate glial scars formation in vivo. To achieve stable and robust delivery of HGF, we transplanted HGF overexpressing-mesenchymal stem cells (MSCs) into the hemisection spinal cord injury site at T8 level (Fig. 6A). After hemisection spinal injury, we immediately implanted gelfoams soaked with PΒS (control group), human bone marrow derived MSCs (MSC alone), and MSCs transduced with HGF gene (HGF-MSC).
Immunohistochemistry was performed to detect surviving grafted cell at 2 weeks after surgery. GFP immunoreactivity was used to detect MSCs because enhanced GFPs were incorporated into MSCs. MSCs were also detected by human specific mitochondria antibodies. Spinal cord tissue of control group (PBS) did not show any immunoreactivity for eGFP or human mitochondria (Fig. 6B). In animals with MSCs alone or HGF-MSCs, eGFP and human mitochondria positive grafted cells were detected inside gelfoam as well as spinal cord parenchyme surrounding the lesion (Fig 6C-F). Thus, grafted MSCs were found to survive for at least 2 weeks post transplantation in both gelfoam around circumference of lesion.
We next examined if HGF overexpressing MSCs were capable of secreting HGF outside of cells in vitro. ELISA analysis of supernatants obtained from culture for different duration showed that the level of HGF secretion increased as the duration of culture was prolonged (Table 3). In contrast, the amount of HGF in supernatants from MSC alone cultures was
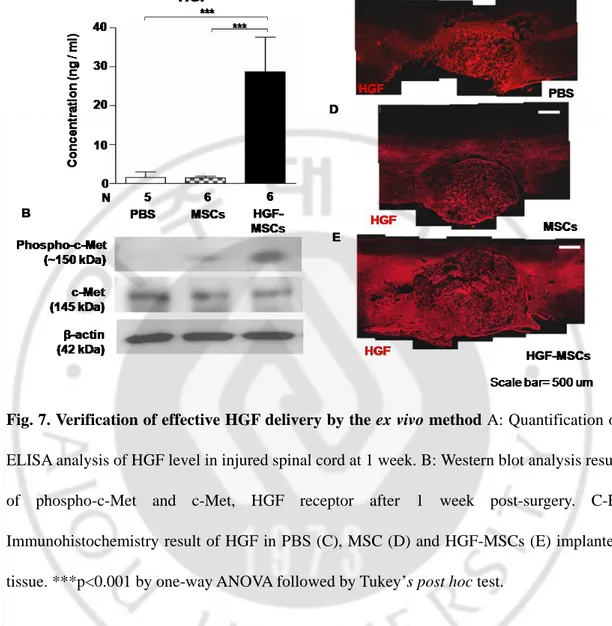
almost negligible. To verify adequate delivery of HGF using the ex vivo approach, animals were sacrificed 1 week after injury, and the level of HGF was measured in the spinal cord tissue around the lesion site. The level of HGF in the spinal cord with HGF-MSCs was 14 times higher than that of control or MSC group (Fig. 7A). This result indicated that the ex
vivo delivery of HGF using MSCs successfully delivered HGF to the injured spinal cord
tissue. Immunohistochemistry using anti-HGF antibodies was also performed to detect presence of HGF in the injured spinal cord tissue (Fig 7C-E). Confocal image showed diffuse HGF immunoreactivity surrounding hemisected injury site in HGF-MSCs group (Fig 7E). Partial expression of PΒS and MSCs groups might be due to endogenous expression of HGFs (Fig. 7C, D).
Binding of HGF to the c-Met receptor tyrosine kinase (RTK) triggers receptor dimerization, which is originally disulfide linked α-β heterodimeric RTK, and phosphorylation on multiple residues (Ma et al., 2003). To determine whether c-Met signaling was effectively induced by the ex vivo approach, phosphorylation of c-Met was measured using phospho c-Met specific antibodies can detect phosphorylation of Tyr 1230, 1234, and 1235 (Fig. 7B). Expression of c-Met receptor was detected to a similar extent in all three groups. Apparent phosphorylation of c-Met was observed in HGF-MSC group, and c-Met phosphorylation was also detected in MSC alone group to a much smaller extent (Fig. 7B).
Fig. 6. Ex vivo delivery of HGF gene using human mesenchymal stem cells as carriers A: Schematic illustration of hemisection spinal cord injury and transplantation of gelfoams with PBS, MSCs alone, and HGF-MSCs. B-E: Results of immunohistochemistry analysis after 1 week post-surgery. B: No cells that were reactive to GFP or human mitochondrial antibodies were detected in animals with PBS soaked gelfoam. C: GFP positive MSCs are readily observed inside the gelfoam. D: GFP and human mitochondria immunofluorescence signals in spinal cord tissue of lesion boundary. E: High magnification fluorescence signal in square of D.
Fig. 7. Verification of effective HGF delivery by the ex vivo method A: Quantification of ELISA analysis of HGF level in injured spinal cord at 1 week. B: Western blot analysis result of phospho-c-Met and c-Met, HGF receptor after 1 week post-surgery. C-E: Immunohistochemistry result of HGF in PΒS (C), MSC (D) and HGF-MSCs (E) implanted tissue. ***p<0.001 by one-way ANOVA followed by Tukey’s post hoc test.
Table 3. HGF concentration in cultured supernatant by culture period condition
Sample HGF concentration (ng/ml)
MSC 3day culture sup (10 MOI) 0.002 HGF 3day culture sup (10 MOI) 2.401 HGF 1day culture sup (10 MOI) 0.599 HGF 2day culture sup (10 MOI) 1.307 HGF 5day culture sup (10 MOI) 17.967
Table 4. Experimental groups and treatment number in vivo
Western blot (neurocan), ELISA (TGFβ1 and TGFβ2) - 1week
groups number
PΒS 5
MSC 6
HGF-MSC 6
Immunohistochemistry (GFAP, CS-56, HGF, eGFP, and Hu-Mito) – 2 weeks
groups number
PΒS 3
MSC 4
E. HGF inhibits astrocytic activation and CSPGs production after spinal cord injury. We sought to investigate whether glial scars formation is influenced by HGF treatment and production of chondroitin sulfate proteoglycans is reduced by HGF treatment after spinal cord injury. First, we examined whether astrocytic hypertrophy in vivo was reduced by HGF as was shown in vitro. At 2 weeks post-injury, glial limitance was formed around the boundary of the hemisection lesion and GFAP expression was increased along the glial limitance (Fig. 8A). Intensity of GFAP immunoreactivity was dramatically suppressed in HGF-MSCs group compared to both PΒS (A) and MSCs (B) groups (Fig. 8A-C). Quantification data showed that transplantation of HGF-MSCs reduced GFAP intensity by more than 50% (Fig. 8D).
In vitro results suggested that HGF prevented astrocytic activation by inhibiting
secretion of TGFβ1 and β2 from reactive astrocytes. To address this issue, we explore whether HGF treatment reduces production of TGFβs in vivo using ELISA analysis (Fig. 9A-B). We found that production of endogenous TGFβ1 was significantly inhibited by transplantation MSCs and HGF-MSCs (Fig. 9A) and the decrease was significantly only by HGF-MSCs transplantation (Fig. 9B).
We next examined the production and deposition of CSPGs. It is well known that CSPGs produced from reactive astrocytes is accumulated in perineuronal nets (PNN) near epicenter after spinal cord injury (Jones et al., 2003). Accumulation of CSPGs was detected by immunoreactivities against GAG sugar chains using CS-56 antibody (Fig. 10A-D). We found that CS-56 immunoreactivity was strongly upregulated in perineuronal net in PΒS implanted group (Fig. 10A-C), indicating that GAGs chains of CSPGs were densely
deposited around the lesion. Transplantation of HGF-MSC markedly inhibited accumulation of CSPGs (Fig. 10C). Transplantation of MSC alone also seemed to decrease CS-56 immunoreactivity (Fig. 10B). Quantification results from CSPG intensity at 3 points that are rostral, caudal and contra lateral was significantly reduced the CS-56 intensity per unit area by both HGF-MSCs and MSCs implantation (Fig. 10D). We also analyzed expression of core proteins of CSPGs by western blot. Neurocan, one of CSPG species, is expressed in injured spinal cord and reaches a peak level at 2 weeks after injury (Jones et al., 2003). Protein homogenates were pretreated with chondroitinase ABC to remove GAGs chains from core protein. The removal of GAG chains allowed visualization of discrete neurocan band at the position expected from its expected molecular weight. We found that HGF-MSCs transplantation reduced intensity of neurocan signals compared to control and MSC alone groups at all the three expected molecular weights (130 kDa, 160 kDa and 240 kDa) (Fig 10E, F). Collectively, these results provide supporting evidence that HGF prevents astrocytic activation and production of CSPGs.
Fig. 8. Reduction of astrocytic scars by transplantation of HGF overexpressing mesenchymal stem cells A-C: Representative images of GFAP stained longitudinal spinal cord tissue sections from animals with PBS (A), MSCs alone (B), and HGFMSCs (C). D: Quantification of GFAP intensity from regions of interests described in methods section. **p<0.01, and ***p<0.001 by one-way ANOVA followed by Tukey’s post hoc test.
Fig. 9. Regulation of TGFβ1 and TGFβ2 level in glial scars by transplantation of HGF-MSCs A-B: Endogenous level of TGFβ1 (A) and TGFβ2 (B) by ELISA analysis. *p<0.05, and ***p<0.001 by one-way ANOVA followed by Tukey’s post hoc test.
Fig. 10. Reduction of CSPGs production by HGF-MSCs in spinal cord injury model A-C: Representative images of CS-56 immunohistochemistry in longitudinal spinal cord sections at 2 weeks post injury in PΒS implantation group (A), MSCs alone implantation group (B), and HGF-MSCs implantation group (C). D: Quantification of CS-56 intensity. E: Western blot analysis result of neurocan. Three bands corresponding to intact (240kDa), N-terminal (160kDa), and C-N-terminal neurocan (130kDa) are shown up. β-actin signal was used as a loading control of tissue lysates. F: Quantification of thickness from 3 bands of 3 sizes. **p<0.01, and ***p<0.001 by one-way ANOVA followed by Tukey’s post hoc test.
Ⅳ. DISCUSSION
In this study, we showed that HGF can prevent astrocytic activation and suppress production of chondroitin sulfate proteoglycans. We further showed that HGF could regulate astrocytic activation via decreasing the level of TGFβs. These effects of HGF on regulation of glial scars were demonstrated both in cultured primary astrocytes and in vivo spinal cord injury model. Our data showed that HGF treatment dramatically reduced morphological changes of astrocytes induced by TGF treatment (hypertrophy) or IFNγ and IL-1beta (elongated morphology) (Fig. 1, 5). As shown at figure 5A-C, we can observe that IL-1β and IFNγ induced activation is modulated the astrocyte active migration and association and HGF could partially prevent these effects, which is involved in cell dissociation and scatter function of HGF (Gherardi et al., 1993). However, HGF didn’t influence astrocyte proliferation in cultured astrocytes (Fig. 2). A previous study is consistent with our result that HGF does not stimulate proliferation of culture astrocytes (Machide et al., 2000). Intriguingly, HGF also completely blocked mRNA expression of neurocan and phosphacan (Fig. 4) that are differently regulated following spinal cord injury (McKeon et al., 1999; Jones et al., 2003). Immunoblotting and immunostaining results indicated that HGF evidently reduced not only depositions of GAG sugar chains (Fig. 10A-D) but also productions of core protein (Fig. 10E-F), which are actively upregulated following injury (Busch and Silver, 2007). Moreover, mRNA expression of c-Met, HGF receptor, is expressed in all condition using RT-PCR in vitro (Fig 3), especially induced at TGFβ1 treatment including HGF co treatment. Previous studies provided the evidence that c-Met is normally
expressed in astrocytes and increases expression level in response to cytokine treatment (TGFβ1, FGF etc) (Shimazaki et al., 2003). Binding of HGF to the c-Met receptor tyrosine kinase (RTK) triggers receptor dimerization, which is originally disulfide linked α-β heterodimeric RTK, and phosphorylation on multiple residues (Ma et al., 2003). We can detect phosphorylated c-Met expression in HGF-MSC transplantation as well as MSC transplantation (Fig. 7B). We thought that weak expression of phospho-c-Met in MSCs group is phosphorylated by other intracellular signaling proteins. TGFβ are well known factor by glial scars inducer, so we suggested that HGF might prevent secretion of TGFβ and then finally HGF can affect blockage of astrocyte activation. ELISA analysis results are supported our hypothesis that HGF treatment has significantly inhibited the secretion of TGFβ1 and TGFβ2 in vitro (Fig. 5E, F) and in vivo (Fig. 9). These results are indicated that hepatocyte growth factor is a potent factor of glial scars prevention and is may be induced the axonal outgrowth around the lesion site and this is first showed that HGF can prevent glial scars formation.
Glial scars are formed around the lesion after spinal cord injury and act as a barrier to regenerating axonal growth. Glial scars mainly consist of reactive astrocytes, which become hypertrophic and greatly increase their expression of intermediate filament proteins such as vimentin, and glial fibrillary acidic proteins (Rhodes and Fawcett, 2004; Busch and Silver, 2007). For in vitro studies, we used two different astrocytes activation schemes: 1) TGFβ1 (Baghdassarian et al., 1993) and 2) IL-1β and IFNγ (Hewett et al., 1993). HGF treatment was effective in preventing GFAP upregulation in TGFβ conditions (Fig. 1, 5). We demonstrated these effects also in in vivo hemisection spinal cord injury model. Thus, these results indicate
that HGFs are effective suppression of GFAP upregulation in response to cytokines or in vivo injury conditions.
Reactive astrocyte inhibits axonal regeneration by producing growth-inhibitory components such as CSPGs (McKeon et al., 1991; Jones et al., 2003). An important implication of our finding is that HGF could block or reduce the production of chondroitin sulfate proteoglycans. CSPGs, such as neurocan, brevican, phosphacan, and NG2, are major elements of the glial scars because secreted CSPGs from reactive astrocyte are accumulated in extracellular matrix (ECM). During the past few years, it has been shown that elimination CSPGs by chondroitinase is effective to improve functional recovery after SCI. In this study, we first found that HGFs decreased the RNA expression of neurocan and phosphacan (Fig. 4), and the deposition of CSPG around the lesion site (Fig. 10) was markedly reduced by transplantation of HGF overexpressing MSCs.
Based on these results, we propose that HGF can modulation of GFAP expression and CSPG production in astrocytes. These effects might be mediated by regulation of TGFβ1 level (Bradbury et al., 2002; Tester and Howland, 2008), which was demonstrated in internal organs such the live and kidney. TGFβ1 is secreted from reactive astrocyte, reactive microglia, and injured neurons, and plays a very important role in constructing the glial scars (Liu and Yang, 2006). Previous studies attempted to reduce glial scars using neutralizing antibodies against TGFβ1 and TGFβ2 (Logan et al., 1994; Buss et al., 2008). Moreover TGFβ1 regulated the production of CSPGs (Logan et al., 1999; Moon and Fawcett, 2001). As expected from our hypothesis, reduction of TGFβ1 and β2 secretion was observed following HGF treatment in vivo and in vitro (Fig. 5, 9). Interestingly, MSC transplantation
is significantly inhibited the secretion of TGFβ1 in vivo (Fig. 9). Zhao et al (2008). already showed that TGFβ1 is decreased following MSC treatment (Asher et al., 2000; Smith and Strunz, 2005). Recently, reactive astrocytes were dramatically inhibited by rapamycin (Zhao et al., 2008a), which is antagonist of mTOR signaling that is serine/threonine kinase and a key regulator of cell size and proliferation downstream of growth factor receptors (Hara et al., 2002). This finding suggests that intracellular signaling of HGF-c-Met system might be somehow related with mTOR signaling. In addition, HGF-c-Met induced delayed STAT3 phosphorylation (Sabatini, 2006; Wullschleger et al., 2006; Bai et al., 2007) that is involved in TGFβ1 intracellular singling (Yin et al., 2008; Lee et al., 2009). Further studies will be required to clarify detailed mechanism by which HGF arrests the secretion TGFβ.
Recent studies have shown that HGF can be therapeutically utilized for various neurological disease conditions. As in our study, HGF overexpressing MSCs were transplanted in stroke animal model to repair the ischemia (Zhao et al., 2008b). They described that the HGF-MSCS were more powerful than MSC cell transplantation alone. Furthermore, a recent study by Kitamura et al. (2007) demonstrated that HGF expressing replication-incompetent herpes simplex virous-1 (HSV-1) vector promoted endogenous repair and functional recovery after spinal cord injury (Zhao et al., 2006), although they did not look into the effects of HGF on glial scars formation. Versatile effects of HGF were actively studied in anti-tumor therapy (Kitamura et al., 2007) and anti-inflammatory inhibition effects (Matsumoto and Nakamura, 2003). HGF was also tested in transgenic mouse ALS model (Kadoyama et al., 2007). In this model, HGF played a role of preventing motoneuronal death and reducing microglia migration following inhibition of pro-apoptotic
protein activation. This study also suggested that secreted HGFs from motoneurons were uptaken by astrocytes and then astrocytes influenced suppression of gliosis via downregulated IL-1β secretion. Thus, HGF and its downstream signaling pathway seem to be a promising target for various neurological disorders.
Ⅴ. CONCLUSION
These results indicate that HGF prevents astrocytic activation and inhibits production of chondroitin sulfate proteoglycans after spinal cord injury in vivo and in vitro. In glial scars mimicking astrocyte cultures, HGF prevented astrocyte activation in a dose-dependent manner. HGF evidently reduced production and deposition of chondroitin sulfate proteoglycans around the lesion. We suggest that inhibition of secretion TGFβ1 and TGFβ2 from activated astrocyte is the mechanism by which HGF prevents the astrocytic activation and production of chondroitin sulfate proteoglycans.
REFERENCE
1. Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW: Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J Neurosci 20: 2427-2438, 2000
2. Baghdassarian D, Toru-Delbauffe D, Gavaret JM, Pierre M: Effects of transforming growth factor-beta 1 on the extracellular matrix and cytoskeleton of cultured astrocytes. Glia 7: 193-202, 1993
3. Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, Jiang Y: Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science 318: 977-980, 2007
4. Border WA, Noble NA: Transforming growth factor beta in tissue fibrosis. N Engl J Med 331: 1286-1292, 1994
5. Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB: Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416: 636-640, 2002
6. Brahmachari S, Fung YK, Pahan K: Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J Neurosci 26: 4930-4939, 2006
7. Busch SA, Silver J: The role of extracellular matrix in CNS regeneration. Curr Opin
Neurobiol 17: 120-127, 2007
8. Buss A, Pech K, Kakulas BA, Martin D, Schoenen J, Noth J, Brook GA: TGF-beta1 and TGF-beta2 expression after traumatic human spinal cord injury. Spinal Cord 46: 364-371, 2008
9. Caggiano AO, Zimber MP, Ganguly A, Blight AR, Gruskin EA: Chondroitinase ABCI improves locomotion and bladder function following contusion injury of the rat spinal cord. J Neurotrauma 22: 226-239, 2005
10. Ebens A, Brose K, Leonardo ED, Hanson MG, Jr., Bladt F, Birchmeier C, Barres BA, Tessier-Lavigne M: Hepatocyte growth factor/scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron 17: 1157-1172, 1996
11. Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV: Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci 24: 2143-2155, 2004
12. Fitch MT, Doller C, Combs CK, Landreth GE, Silver J: Cellular and molecular mechanisms of glial scarsring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci 19:
8182-8198, 1999
13. Galtrey CM, Fawcett JW: The role of chondroitin sulfate proteoglycans in regeneration and plasticity in the central nervous system. Brain Res Rev 54: 1-18, 2007
14. Garzotto D, Giacobini P, Crepaldi T, Fasolo A, De Marchis S: Hepatocyte growth factor regulates migration of olfactory interneuron precursors in the rostral migratory stream through Met-Grb2 coupling. J Neurosci 28: 5901-5909, 2008
15. Gherardi E, Sharpe M, Lane K, Sirulnik A, Stoker M: Hepatocyte growth factor/scatter factor (HGF/SF), the c-met receptor and the behaviour of epithelial cells. Symp Soc
Exp Biol 47: 163-181, 1993
16. Gomes FC, Sousa Vde O, Romao L: Emerging roles for TGF-beta1 in nervous system development. Int J Dev Neurosci 23: 413-424, 2005
17. Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K: Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110: 177-189, 2002
18. Hewett SJ, Corbett JA, McDaniel ML, Choi DW: Interferon-gamma and interleukin-1 beta induce nitric oxide formation from primary mouse astrocytes. Neurosci Lett 164: 229-232, 1993
19. Hudgins SN, Levison SW: Ciliary neurotrophic factor stimulates astroglial hypertrophy in vivo and in vitro. Exp Neurol 150: 171-182, 1998
20. John GR, Lee SC, Brosnan CF: Cytokines: powerful regulators of glial cell activation.
Neuroscientist 9: 10-22, 2003
21. Jones LL, Tuszynski MH: Spinal cord injury elicits expression of keratan sulfate proteoglycans by macrophages, reactive microglia, and oligodendrocyte progenitors.
J Neurosci 22: 4611-4624, 2002
22. Jones LL, Margolis RU, Tuszynski MH: The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol 182: 399-411, 2003
23. Kadoyama K, Funakoshi H, Ohya W, Nakamura T: Hepatocyte growth factor (HGF) attenuates gliosis and motoneuronal degeneration in the brainstem motor nuclei of a transgenic mouse model of ALS. Neurosci Res 59: 446-456, 2007
24. Kim TH, Mars WM, Stolz DB, Petersen BE, Michalopoulos GK: Extracellular matrix remodeling at the early stages of liver regeneration in the rat. Hepatology 26: 896-904, 1997
Shibata S, Funakoshi H, Miyatake S, Coffin RS, Nakamura T, Toyama Y, Okano H: Hepatocyte growth factor promotes endogenous repair and functional recovery after spinal cord injury. J Neurosci Res 85: 2332-2342, 2007
26. Kobayashi E, Sasamura H, Mifune M, Shimizu-Hirota R, Kuroda M, Hayashi M, Saruta T: Hepatocyte growth factor regulates proteoglycan synthesis in interstitial fibroblasts. Kidney Int 64: 1179-1188, 2003
27. Krasnoselsky A, Massay MJ, DeFrances MC, Michalopoulos G, Zarnegar R, Ratner N: Hepatocyte growth factor is a mitogen for Schwann cells and is present in neurofibromas. J Neurosci 14: 7284-7290, 1994
28. Lee BS, Park M, Cha HY, Lee JH: Hepatocyte growth factor induces delayed STAT3 phosphorylation through interleukin-6 expression. Cell Signal 21: 419-427, 2009
29. Levison SW, Hudgins SN, Crawford JL: Ciliary neurotrophic factor stimulates nuclear hypertrophy and increases the GFAP content of cultured astrocytes. Brain Res 803: 189-193, 1998
30. Liu Y: Hepatocyte growth factor in kidney fibrosis: therapeutic potential and mechanisms of action. Am J Physiol Renal Physiol 287: F7-16, 2004
213-217, 2006
32. Liu Y, Yang J: Hepatocyte growth factor: new arsenal in the fights against renal fibrosis?
Kidney Int 70: 238-240, 2006
33. Logan A, Green J, Hunter A, Jackson R, Berry M: Inhibition of glial scarsring in the injured rat brain by a recombinant human monoclonal antibody to transforming growth factor-beta2. Eur J Neurosci 11: 2367-2374, 1999
34. Logan A, Berry M, Gonzalez AM, Frautschy SA, Sporn MB, Baird A: Effects of transforming growth factor beta 1 on scar production in the injured central nervous system of the rat. Eur J Neurosci 6: 355-363, 1994
35. Ma PC, Maulik G, Christensen J, Salgia R: c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev 22: 309-325, 2003
36. Machide M, Kamitori K, Kohsaka S: Hepatocyte growth factor-induced differential activation of phospholipase cgamma 1 and phosphatidylinositol 3-kinase is regulated by tyrosine phosphatase SHP-1 in astrocytes. J Biol Chem 275: 31392-31398, 2000
37. Maina F, Klein R: Hepatocyte growth factor, a versatile signal for developing neurons.
38. Matsumoto K, Nakamura T: Hepatocyte growth factor: molecular structure, roles in liver regeneration, and other biological functions. Crit Rev Oncog 3: 27-54, 1992
39. Matsumoto K, Nakamura T: NK4 (HGF-antagonist/angiogenesis inhibitor) in cancer biology and therapeutics. Cancer Sci 94: 321-327, 2003
40. McKeon RJ, Jurynec MJ, Buck CR: The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scars. J
Neurosci 19: 10778-10788, 1999
41. McKeon RJ, Schreiber RC, Rudge JS, Silver J: Reduction of neurite outgrowth in a model of glial scarsring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci 11: 3398-3411, 1991
42. Moon LD, Fawcett JW: Reduction in CNS scar formation without concomitant increase in axon regeneration following treatment of adult rat brain with a combination of antibodies to TGFbeta1 and beta2. Eur J Neurosci 14: 1667-1677, 2001
43. Nakamura T, Sakata R, Ueno T, Sata M, Ueno H: Inhibition of transforming growth factor beta prevents progression of liver fibrosis and enhances hepatocyte regeneration in dimethylnitrosamine-treated rats. Hepatology 32: 247-255, 2000
Shimizu S: Molecular cloning and expression of human hepatocyte growth factor.
Nature 342: 440-443, 1989
45. Ohya W, Funakoshi H, Kurosawa T, Nakamura T: Hepatocyte growth factor (HGF) promotes oligodendrocyte progenitor cell proliferation and inhibits its differentiation during postnatal development in the rat. Brain Res 1147: 51-65, 2007
46. Pekny M, Nilsson M: Astrocyte activation and reactive gliosis. Glia 50: 427-434, 2005
47. Reilly JF, Maher PA, Kumari VG: Regulation of astrocyte GFAP expression by TGF-beta1 and FGF-2. Glia 22: 202-210, 1998
48. Rhodes KE, Fawcett JW: Chondroitin sulphate proteoglycans: preventing plasticity or protecting the CNS? J Anat 204: 33-48, 2004
49. Ribotta MG, Menet V, Privat A: Glial scars and axonal regeneration in the CNS: lessons from GFAP and vimentin transgenic mice. Acta Neurochir Suppl 89: 87-92, 2004
50. Rohl C, Armbrust E, Kolbe K, Lucius R, Maser E, Venz S, Gulden M: Activated microglia modulate astroglial enzymes involved in oxidative and inflammatory stress and increase the resistance of astrocytes to oxidative stress in vitro. Glia 56: 1114-1126, 2008
51. Rolls A, Shechter R, Schwartz M: The bright side of the glial scars in CNS repair. Nat
Rev Neurosci 10: 235-241, 2009
52. Sabatini DM: mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 6: 729-734, 2006
53. Sandvig A, Berry M, Barrett LB, Butt A, Logan A: Myelin-, reactive glia-, and scar-derived CNS axon growth inhibitors: expression, receptor signaling, and correlation with axon regeneration. Glia 46: 225-251, 2004
54. Shimamura M, Sato N, Sata M, Wakayama K, Ogihara T, Morishita R: Expression of hepatocyte growth factor and c-Met after spinal cord injury in rats. Brain Res 1151: 188-194, 2007
55. Shimazaki K, Yoshida K, Hirose Y, Ishimori H, Katayama M, Kawase T: Cytokines regulate c-Met expression in cultured astrocytes. Brain Res 962: 105-110, 2003
56. Silver J, Miller JH: Regeneration beyond the glial scars. Nat Rev Neurosci 5: 146-156, 2004
57. Smith GM, Strunz C: Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia 52: 209-218, 2005
58. Tang X, Davies JE, Davies SJ: Changes in distribution, cell associations, and protein expression levels of NG2, neurocan, phosphacan, brevican, versican V2, and tenascin-C during acute to chronic maturation of spinal cord scar tissue. J Neurosci
Res 71: 427-444, 2003
59. Tester NJ, Howland DR: Chondroitinase ABC improves basic and skilled locomotion in spinal cord injured cats. Exp Neurol 209: 483-496, 2008
60. Tom VJ, Houle JD: Intraspinal microinjection of chondroitinase ABC following injury promotes axonal regeneration out of a peripheral nerve graft bridge. Exp Neurol 211: 315-319, 2008
61. Wullschleger S, Loewith R, Hall MN: TOR signaling in growth and metabolism. Cell 124: 471-484, 2006
62. Yamamoto Y, Livet J, Pollock RA, Garces A, Arce V, deLapeyriere O, Henderson CE: Hepatocyte growth factor (HGF/SF) is a muscle-derived survival factor for a subpopulation of embryonic motoneurons. Development 124: 2903-2913, 1997
63. Yan H, Rivkees SA: Hepatocyte growth factor stimulates the proliferation and migration of oligodendrocyte precursor cells. J Neurosci Res 69: 597-606, 2002